Concept of crystal field theory. Models of chemical bonding. Crystal field theory. Low- and high-spin complexes
Weak field strong field
Middle field
Frac34;¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾® Δo
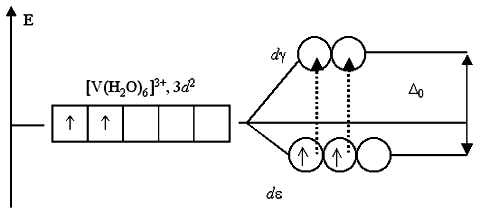
Weak field ligands with elements of the 3d series form high-spin complexes, and strong-field ligands form low-spin complexes. The difference between them affects the electronic structure of the complexes only for configurations d 4 – d 7:
3+ d 5 3– d 5
high-spin complex low-spin complex
H 2 O – weak field ligand CN – – strong field ligand
Low-spin complexes are always more stable than high-spin complexes. Medium-field ligands, depending on conditions (charge and nature of the central atom), can form both high-spin and low-spin complexes.
Example. Based on TCP, make an assumption about the electronic structure of hexaammine cobalt(II) (Δo = 21600 cm–1, P = 21000 cm–1) and hexaammine cobalt(III) ions (Δo = 9500 cm–1, P = 22500 cm–1).
Ammonia is a medium-field ligand and, depending on the degree of oxidation of the metal, can form both high-spin and low-spin complexes. Let us find out which complexes will be energetically more stable for cobalt(II) and cobalt(III). To do this, compare the ESC of each ion in a strong and weak field:
(a) 3+, d 6

strong field weak field
ESKP (strong field) = –6´(2/5)Δo + 2P = –6´(2/5) ´21600 + 2´21000 = –9840 cm –1
ESKP (weak field) = –4´(2/5)Δo + 2´(3/5)Δo = –4´(2/5) ´21600 + 2´(3/5) ´21600 = –8640 cm – 1
The energy gain is greater in the case of a low-spin complex.
(b) 2+ , d 7

strong field weak field
ESKP (strong field) = –6´(2/5)Δo + 1´(3/5)Δo + P = –6´(2/5)´9500 + 1´(3/5) ´9500 + 22500 = 7900 cm–1
ESKP (weak field) = –5´(2/5)Δo + 2´(3/5)Δo = –5´(2/5) ´9500 + 2´(3/5) ´9500 = –7600 cm – 1
The energy gain is greater in the case of a high-spin complex.
Thus, the 3+ ion is low-spin and the 2+ ion is high-spin.
The ESC increases with increasing Δo, however, it is different for high-spin and low-spin states (Fig. 1.28. The dependence of the ESC for high-spin and low-spin complexes with configuration d 6 on the value Δo = 10Dq. The region in which the existence of both states is possible is shaded). The region near the intersection point of these two lines corresponds to complexes that can exist in both high-spin and low-spin states.
An example is the iron(II) thiocyanate complex with 1,10-phenanthroline, which is high-spin (paramagnetic) at low temperatures, and low-spin (diamagnetic) at elevated temperatures (M. Marchivie, P. Guionneau, J. A. K. Howard, G. Chastanet, J.-F. Letard, A. E. Goeta, D. Chasseau, J. Am. Chem. Soc., 2002, v. 124, p. 194). The change in multiplicity is accompanied by a change in interatomic distances and the geometry of the coordination environment: the low-spin complex is a regular octahedron, and the high-spin complex is a distorted one. The reverse transition to the high-spin state is possible under the influence of high pressures or radiation. Currently, several dozen such systems are known.
Speaking about the σ-donor and π-acceptor properties of the ligand, we went beyond the TCP, using the approaches of the molecular orbital method as applied to complex compounds (Volume 1). Let us recall that the picture of the splitting of d-orbitals is a fragment of the general scheme of molecular orbitals in an octahedral complex, where t 2g orbitals are considered as non-bonding, and e g - as antibonding (Fig. Volume 1).
The formation of bonds in an octahedral complex without π-bonding involves the s-, p- and d-orbitals of the metal and one orbital from each ligand. From 15 atomic orbitals, 15 molecular orbitals are formed, six of them (a 1 g, t 1 u, e g (footnote: the letter in the designation of orbitals indicates the degree of their degeneracy: t - three times degenerate, e - doubly degenerate, a - non-degenerate, and the presence of a center of symmetry: g - symmetrical, u - asymmetrical)) σ-bonding, three (t 2 g) - non-bonding, and six (e g *, t 1 u *, a 1 g *) σ-loosening. Bonding orbitals are closer in energy to the ligand orbitals, while nonbonding orbitals are localized predominantly on the metal atom. The energy d xy , d xz , d yz (t 2 g) of the metal orbitals practically does not change during the formation of the complex.
The presence of a low-energy vacant orbital in the ligand, similar in symmetry to the metal orbitals, leads to a decrease in the energy of t 2g orbitals, practically without affecting eg, thereby increasing Δо (Fig. 1.29. Fragments of the MO diagram for the cobalt(III) complex with σ-donor ligand (a) and σ-donor, π-acceptor ligand (b)).
Jahn-Teller effect. In 1937, Yang and Teller proved the theorem according to which any nonlinear molecule in a degenerate electronic state is unstable and spontaneously undergoes a distortion that lowers its symmetry and leads to the removal of degeneracy. The theorem predicts only the very fact of removing degeneracy, but does not indicate how it will be removed. Based on this theorem, the distortion of the octahedral geometry of a number of complexes was explained, and the very fact of the presence of such a distortion was called the Jahn-Teller effect. Let's look at an example. Copper(II) complexes with the d9 configuration, as a rule, do not represent a regular octahedron, but are elongated or compressed along one of the axes (Fig. 1.30. Distortion of octahedral geometry in copper(II) complexes). Let us consider the case of a prolate octahedron. Removal of ligands located along the z axis causes the removal of degeneracy due to a change in the energies of the orbitals. Orbitals directed along the z axis (d xz, d yz, d z 2) interact weaker with the orbitals of the ligands compared to orbitals that do not have a z component (d xy, d x 2 -y 2), and therefore lower their energy. A pair of orbitals of the same symmetry, having a z-component (d xz, d yz), remains degenerate and acquires increased energy. (Fig. 1.31. Change in the energies of d-orbitals when the octahedron is distorted). The Jahn-Teller effect manifests itself most strongly in complexes with unequally filled e g orbitals, that is, with configurations t 2g 3 e g 1 (corresponding to the d 4 ion in a weak field: CrCl 2, K 3 MnF 6) and t 2g 6 e g 3 ( corresponds to the d 9 ion: almost all copper(II) complexes) and t 2g 6 e g 1 (corresponds to the d 7 ion in a strong field, rare, K 3 NiF 6),. An insignificant Jahn-Teller effect is typical for complexes with unequally filled t 2g orbitals, that is, for electronic configurations t 2g 1 (d 1), t 2g 2 (d 2), t 2g 4 (d 4 in a strong field), t 2g 5 (d 5 in a strong field), t 2g 5 e g 1 (d 6 in a weak field), t 2g 5 e g 2 (d 7 in a weak field). Ions with configurations d 3 and d 5 in a weak field, d 3 and d 6 in a strong field, d 8 and d 10 are under no circumstances Jahn-Teller.
The Jahn-Teller effect manifests itself in the inequality of bond lengths in many copper(II) and manganese(III) complexes and in a nonmonotonic change in the stepwise stability constants of the complexes. For example, in anhydrous copper(II) chloride, the copper atom is surrounded by six chlorine atoms, four of which are located at a distance of 0.230 nm, and the other two are located at a distance of 0.295 nm from it.
Copper(II) complexes (Cl 2, (C 6 H 5 SO 3) 2, etc.) are known, consisting of several crystallographically nonequivalent Jahn-Teller ions, each with its own type of distortion, which transform into each other, changing the metal-ligand distance so fast that overall all metal-ligand distances appear to be the same. This case was called dynamic or pulsating Jahn-Teller effect(P. E. M. Wijnands, J. S. Wood, J. Redijk, W. J. A. Maaskant, Inorg. Chem., 1986, 35, 1214) .
The Jahn-Teller effect, however, is not a universal law. Currently, complex ions with a Jahn-Teller configuration are known, which are undistorted octahedra: 4–, 3+.
Splitting in fields with symmetry other than octahedral.
In addition to octahedral ones, there are many known complexes with a different geometry - square-plane, tetrahedral, trigonal-pyramidal, square-pyramidal, linear, etc. The splitting in each of these fields is different than in the octahedron; it is determined by the symmetry of the coordination polyhedron.
Square-planar complexes can be considered as an extreme case of tetragonal distortion of the octahedral geometry, when the ligands located along one of the coordinate axes are removed to infinity (Fig. 1.27b). The designations of the orbitals are shown in the figure. Planar-square complexes are most typical for ions with the electronic configuration d 8 – Ni 2+, Pd 2+, Pt 2+, Au 3+. Their stability increases sharply with increasing Δ, that is, when moving from elements of the 3d series to heavy transition elements. So, for example, if palladium, platinum and gold have almost all complexes with a coordination number of four square, then nickel forms planar-square complexes only with high-field ligands: 2–, Ni(dmg) 2. Nickel(II) complexes with low-field ligands, such as halogens, have a tetrahedral geometry.
Some square-planar transition metal complexes form chains in solid form with bridging ligands, for example Pt-CN-Pt in K 2 Br 0.3, where the platinum atoms are partially in the +4 oxidation state. The high penetrating ability of 5d orbitals ensures their overlap with the formation of a single energy band, and, consequently, metallic conductivity in the direction of the chain. Such molecular complexes are capable of conducting electrical current and are currently being intensively studied.
In a field of tetrahedral symmetry, the orbitals d xy , d yz , d xz have the maximum energy, they are called t 2 -orbitals, and the minimum energy is the orbitals d x 2 –y 2 and d z 2, they are denoted e. Due to the presence of a smaller number of ligands and their different arrangement, the tetrahedral field (Fig. 1.32. Comparison of splittings in the tetrahedral and octahedral fields) turns out to be 2.25 times weaker than the octahedral one: .
Most tetrahedral complexes are high-spin. (Footnote - Several examples of low-spin tetrahedral complexes are known, for example, Cr(N(Si(CH 3) 3) 2 ) 3 NO (chromium(II), d 4 ; D. C. Bradley, Chem. Ber., 1979 , 11, 393); CoL 4, where L is 1-norbornyl (cobalt(IV), d 5; E. K: Brune, D. S. Richeson, K. H. Theopold, Chem. Commun., 1986, 1491)). Maximum stabilization of the tetrahedral environment by the crystalline field is achieved with configurations d 2 (FeO 4 2–, MnO 4 3–) and d 7 (2–). Due to the relatively low stabilization energy, tetrahedral complexes are more often formed by ions with configurations d 0 (TiCl 4, MnO 4 –, CrO 4 2–), d 5 in a weak field (FeCl 4 –) and d 10 (ZnCl 4 2–) with zero ESKP, as well as non-transition metal ions (AlCl 4 –). The formation of tetrahedral complexes compared to octahedral ones is often favored by the steric factor, for example, the ion is more stable than 3–.
Using TCP to explain the stability of complexes. Irving-Williams series. The crystal field theory makes it possible to explain the non-monotonic nature of changes in the energies of the crystal lattice of oxides and halides, stability constants of complexes, etc. The order of change in the hydration energies of doubly charged cations of 3d metals generally coincides with the nature of changes in the ESC in high-spin complexes (Fig. 1.33. Change in the hydration energy of doubly charged cations metals of the 3d series (a) and the change in ESC in high-spin complexes (b)), the stronger the stabilization by the crystal field, the greater the hydration. It is known that the constants of substitution of a water molecule by a weak-field ligand L
2+ + L x– = (2-x)+ + H 2 O
obey the Irving-Williams series: Mn 2+< Fe 2+ < Co 2+ < Ni 2+ < Cu 2+ < Zn 2+ (Рис. 1.34. Зависимость первой константы устойчивости комплекса от природы 3d-металла). Согласно этому ряду, наибольшей устойчивостью обладают комплексы меди(II) и никеля(II). Простейший вариант ЭСКП предсказывает наибольшую устойчивость никелевых комплексов. При этом надо учитывать, что комплексы меди(II) имеют сильно искаженную октаэдрическую геометрию, что вносит существенный вклад в величину константы устойчивости.
Nepheloauxetic effect. It was discovered that the mutual repulsion of d-electrons weakens when the atom is placed in the field of ligands. This effect of the ligand on the d-electrons of the metal atom is called the nepheloauxetic effect from the Greek words νεφελη - cloud and αυξανω - increase. The series of ligands, arranged in order of increasing their influence on the metal orbitals, almost completely corresponds to the spectrochemical series. The reason for the nepheloaxetic effect is the overlap of the d-orbitals of the metal with the orbitals of the ligands, due to which the d-cloud expands in space. The presence of this effect clearly demonstrates the limitations of the simplest electrostatic model - the crystalline field theory, which assumes that lignades are point negative charges.
Ligand field theory. Crystal field theory was developed by Bethe in 1929. Currently, it is widely used in a modified form with corrections for some covalency of the metal-ligand bond. This theory is called ligand field theory. The presence of a covalent contribution changes the energy of the metal orbitals in comparison with that calculated by TCP. The proportion of covalency is taken into account by introducing correction factors that make it possible to equate the experimental values with the calculated ones.
Coloring of complexes.
The color of d-transition element complexes is associated with electron transitions from one d-orbital to another. This is clearly illustrated by the example of the Ti 3+ ion, discussed in the first volume of the textbook. By absorbing energy corresponding to the blue and green parts of the visible spectrum, the only d-electron in the Ti 3+ ion moves to the e g orbital (Fig. 1.35. Spectrum of the 3+ ion). The color of the ion is due to additional colors - red and violet. (Footnote - The attentive reader will notice some asymmetry of the absorption band. It is a consequence of a slight splitting of the t 2g level caused by the Jahn-Teller effect). A diagram showing complementary colors and which is well known to every artist is presented on the second flyleaf of the textbook. The transition energy, expressed in reciprocal centimeters (1000 cm –1 = 12 kJ), corresponds to the splitting parameter Δο - it is most often determined from electronic spectra. Wavelength is inversely proportional to energy:
 .
.
In the case of complexes with a large number of electrons, the spectrum picture becomes more complicated, and additional bands appear in it. This is due to the fact that the excited state t 2g 1 e g 1 can be realized in several ways, depending on which two d-orbitals the electrons are in. For example, a state in which electrons occupy d xy and d x 2 –y 2 orbitals will be higher in energy than a d xy 1 d z 2 1 state due to the greater repulsion of electrons along the x axis. The energy corresponding to the band with the lowest energy is equal to the splitting parameter Δo.
To describe electronic spectra in more detail, it is necessary to introduce some concepts. Let us call any arrangement of electrons at a sublevel a microstate. The number of microstates N, in which n electrons occupy x orbitals, is equal to

Each microstate is characterized by its own values of spin and angular momentum. A set of microstates with identical energies is called term, for example, 3 P, 5 D, 1 S. The digital index indicates multiplicity, which is calculated as:
multiplicity = number of unpaired electrons in the ground state + 1.
The names of the terms are read with an indication of multiplicity: “triplet P”, “quintet D”, “singlet S”. The letter denotes the total angular momentum L of an atom or ion, which is equal to the maximum value of the sum of the angular momenta m l of individual orbitals occupied by electrons. For example, the Ti 3+ ion contains one d-electron, the number of microstates is N = (2´5)!/1!(2´5 – 1)! = 10, L = 2(D) (since for the d-orbital m l = –2, –1, 0, 1, 2, the number of electrons is 1, therefore, the maximum sum m l is equal to the largest value of m l), multiplicity 1 + 1 = 2. Therefore, the ground state term (with the lowest energy) is 2 D. In the case of an ion with an electronic configuration d 2 N = (2´5)!/2!(2´5 – 2)! = 45, L = 3(F) (since for the d-orbital m l = –2, –1, 0, 1, 2, the number of electrons is 2, therefore, the maximum sum of the two largest values is equal to m l), multiplicity 2 + 1 = 3. Consequently, the term of the ground microstate is 3 F. With a different arrangement of two electrons on the d-sublevel, states described by other terms are achieved - 3 P, 1 G, 1 D, 1 S, etc. The relationship between the numerical values of L and the alphabetic symbols is given below:
L = 0 1 2 3 4 5 6 7
Similarly, we can derive the terms of the ground and excited states for other ions of d-elements (Table 1.5.). Please note that the terms for ions with configuration d n and d 10-n are the same.
Table. 1.5.
Terms of the ground and nearest excited states for various configurations of d-electrons.
The terms are split in the octahedral field like orbitals, denoted by similar letters. D terms are split into T 2 g and E g components, like d-orbitals, F terms - into T 1 g, T 2 g and A 2 g, like f-orbitals. The S and P terms are not split at all. The possibilities for electron transitions between different states are limited by selection rules. Thus, in complexes only transitions between states with the same multiplicity are allowed. Each such transition corresponds to a band in the absorption spectrum. As an example, consider the electronic spectrum of complex 3+ (Fig. 1.36. Electronic spectrum of complex 3+). The three bands are due to three electronic transitions: 4 A 2 g ® 4 T 2 g, 4 A 2 g ® 4 T 1 g, 4 A 2 g ® 4 T 1 g (P). The transition with the lowest energy corresponds to the value of the splitting parameter: Δo = 17400 cm–1. The complex absorbs light in the red (17400 cm–1) and blue (23000 cm–1) parts of the visible spectrum and in the near ultraviolet (37800 cm–1), therefore, it has a violet color.
According to Laporte's rule, transitions between states with the same parity, which include s-s, p-p, d-d, f-f transitions, are unlikely, or, in the language of spectroscopy, they are prohibited in octahedral complexes. Forbidden transitions are possible, but occur with low intensity. This is why transition metal salts have a noticeable color only in concentrated solutions. It is many times weaker than the color of permanganate or dichromate, the ions of which do not contain d-electrons.
Laporte's rule is applicable only in the case of complexes that have a center of symmetry. When the octahedron is distorted, the center of symmetry disappears, the Laporte prohibition is lifted, and color appears. For example, the 3+ ion is colorless, but solutions of iron(III) salts are often yellow-orange due to hydrolysis leading to the formation of asymmetric particles with a distorted octahedral environment.
The color of the complexes, in addition to d-d transitions from one metal d-orbital to another (from t 2g to e g in octahedral complexes), is determined by two more factors: transitions from ligand orbitals to metal orbitals (they are called charge transfer) and transitions within the ligand orbitals. These transitions do not fall under Laporte's rule and, therefore, have high intensity.
The charge transfer band is present in the electronic spectrum of any compound, however, in some cases it is in the ultraviolet part of the spectrum and is not perceived by us as color. If the difference between the energies of the ligand orbitals and the metal orbitals is reduced, the charge transfer band falls into the visible part of the spectrum. It is charge transfer that explains the intense color of permanganate, dichromate, mercury sulfide, titanium(IV) peroxo complexes and many other compounds with empty d-orbitals. In some cases, under the influence of light, charge transfer from the orbitals of the ligand to the orbitals of the metal occurs irreversibly, that is, it is accompanied by a chemical process. An example is the photochemical decomposition of silver halides, which is the basis of black and white photography: Ag + Br – ¾® Ag 0 + Br 0 .
In the electronic spectrum of potassium permanganate, four bands are observed, corresponding to transitions of electrons from nonbonding orbitals localized predominantly on the ligand (a 1, t 2 σ orbitals and e, t 1, t 2 π orbitals) to e*, t2'' antibonding orbitals orbitals localized on the metal atom ((Fig. 1.37. Energy diagram of the tetrahedral ion MnO 4 - with π-bonding. Electron transitions are shown by arrows):
ν 1 , Mn(e*) ¾ O(t 1) 17700 cm –1
ν 2 , Mn(t 2 '') ¾ O(t 1) 29500 cm –1
ν 3 , Mn(e*) ¾ O(t 2) 30300 cm –1
ν 4 , Mn(t 2 '') ¾ O(t 2) 44400 cm –1
The band with the lowest energy falls in the visible part of the spectrum (λ = 107/17700 = 565 nm), which corresponds to the absorption of green light and the transmission of crimson-red light.
3. Mechanisms of reactions involving complex compounds.
The vast majority of chemical processes occur as a sequential chain of some elementary stages, and the reaction equation carries only information about the main end products of the reaction. This sequence of elementary transformations on the way from starting substances to products is called a mechanism. Intermediate, usually unstable compounds through which the path from reactants to products runs are called intermediates. Any intermediate has a certain lifetime, usually extremely short, up to 10 -14 s. On the energy profile of the reaction it corresponds to a minimum (Fig. a) (Fig. 1.38. Energy profiles of a reaction proceeding through: (a) intermediate, (b) transition state.). As a rule, intermediates can be detected in a reaction mixture by spectral methods, and only in rare cases can they be isolated in individual form. Therefore, the main information about the reaction mechanism is usually obtained through studying its kinetics - determining rate constants and calculating activation parameters (enthalpy, entropy, volume). In this case, the mechanism is a model that is in accordance with the kinetic data, a model that can be improved, modified, revised.
In some reactions, intermediates are not formed, and the transition from reactants to products occurs sequentially - one of the atoms is gradually removed, and the other approaches. In this case, the reaction is said to proceed through transition state or activated complex. It corresponds to a maximum in the energy profile of the reaction (Fig. B).
Addition: Labile and inert complexes
The thermodynamic stability of a particle is determined by the change in the Gibbs energy for the reaction of its dissociation, or by the value of the stability constant of this process. Kinetic stability shows how quickly a given particle interacts with other particles or undergoes decay. A chemical particle is considered inert, if it reacts with a half-life of more than 1 minute. Particles that react at a higher rate are called labile. It must be remembered that kinetic and thermodynamic stability do not depend on one another, that is, the same substance can have a high stability constant and at the same time be inert, or, conversely, labile. Some such examples are given in Table 1.6.
Table 1.6. Stability constants and rates of ligand substitution in cyano-complexes of some metals.
Henry Taube showed the connection between the kinetic stability of octahedral complexes and the electronic configuration of the central ion in the octahedral field. According to Taube, the following complexes are labile:
· possessing at least one vacant t 2g orbital - they can use it in reactions according to the associative (A, I a) mechanism, or
· having at least one electron in the e g orbital - this promotes the reaction by the dissociative (D, I d) mechanism, because Removing an electron from the e g orbital lowers the energy of the transition state.
Thus, octahedral complexes of chromium(III) (t 2g 3), low-spin complexes of iron(II) (t 2g 6) and iron(III) (t 2g 5), as well as complexes of 4d-, 5d-transition elements are classified as inert with the number of d-electrons more than two.
END OF ADDENDUM
A unified classification of inorganic reactions has not yet been developed. Conventionally, we can propose the following scheme (Fig. 1.39. Scheme illustrating the classification of inorganic reactions):
1) Reactions of substitution, addition or elimination of ligands affect a change in the coordination sphere of the metal,
2) Redox reactions are associated with a change in the electronic configuration of the metal, but do not affect its coordination environment,
3) Reactions of coordinated ligands involve a change in the ligand without changing the coordination sphere of the complex.
Substitution reactions. In a broad sense, substitution reactions mean the processes of replacing some ligands in the coordination sphere of a metal with others. Such reactions can occur either with or without a change in the oxidation state. Following the above classification, we will use this term only in relation to reactions that occur without a change in oxidation states.
The classification of substitution reactions in inorganic chemistry was developed by Langford and Gray. It is based on the definition of the so-called limiting mechanism, and not on the description of a specific mechanism. First, the stoichiometric mechanism is determined, and then the internal one. Stoichiometric mechanism is a sequence of elementary stages in the transition from starting substances to products. It can be dissociative (D), associative (A) and exchange (reciprocal exchange, I). Dissociative and associative processes represent, as it were, two limiting cases, directly opposite to one another. Both processes occur in two stages through the formation of an intermediate.
Dissociative (D)
The process is two-stage, in the limiting case it proceeds through an intermediate with a reduced concentration:
ML 6 + L, + Y ¾® ML 5 Y
Associative (A)
The process is two-stage, characterized by the formation of an intermediate with an increased concentration:
ML 6 + Y, ¾® ML 5 Y + L
Mutual exchange (I)
Most exchange reactions proceed through this mechanism. The process is one-stage and is not accompanied by the formation of an intermediate. In the transition state, the reagent and the leaving group are associated with the reaction center, enter its nearest coordination sphere, and during the reaction one group is displaced by another, an exchange of two ligands occurs:
ML 6 + Y ML 5 Y + L.
The transition state is either an outer-sphere complex or, in the case of charged ligands, an ion pair MX 5 L + Y - .
Internal mechanism (a or d) characterizes the process of ligand substitution at the molecular level. It shows which of the two processes - the formation or rupture of a bond in the transition state - is limiting. If the reaction rate is determined by the formation of a bond between the reaction center and the reagent, we speak of associative activation. Otherwise, when the limiting factor is the rupture of the connection between the reaction center and the leaving group, the process proceeds with dissociative activation. Turning to the stoichiometric mechanism, it is easy to notice that the dissociative process always corresponds to dissociative activation, and the associative process always corresponds to associative activation, that is, the concept of an internal mechanism turns out to be informative only in the case of a mutual exchange mechanism - it can occur with both dissociative (I d) and associative (I a) activation. In the case of the reciprocal exchange mechanism with associative activation (Ia), the reaction rate depends on the nature of Y. In the transition state, the metal atom is tightly bound to both the leaving group and the attacking nucleophile. An example is the process of replacing a chlorine atom with bromine and iodine in a platinum complex with diethylenetriamine (dien):
Y - ¾¾® + + Cl -
Y = Br, I velocities vary greatly.
In the case of the reciprocal exchange mechanism with dissociative activation (I d), the reaction rate does not depend on the nature of the reagent Y. The attacking and leaving groups in the transition state are weakly bound to the central ion. This mechanism is used to replace water with amine in aqua complexes of many transition metals, for example, nickel:
2+ + Y ¾¾® 2+ + H 2 O
Y = NH 3 , py velocities are close.
The study of the mechanisms of substitution reactions in complexes of many metals is only in the initial stage. Comprehensive information has been obtained only for square-planar complexes of platinum and octahedral complexes of chromium(III) and cobalt(III). It can be considered firmly established that in platinum(II) complexes, substitution occurs according to the associative mechanism (A, Ia) through an intermediate or transition state in the form of a trigonal bipyramid. Octahedral cobalt(III) complexes react dissociatively (D, I d mechanisms). Specific examples of such reactions will be considered when describing the chemistry of these elements.
Redox reactions. Most redox processes are a complex combination of individual elementary stages, each of which involves the transfer of one or, much less frequently, two electrons. Simultaneous transfer of a larger number of electrons in solutions is impossible.
Single-electron transfer can occur through one of two mechanisms: outer-sphere, that is, by tunneling, or inner-sphere, through a bridging ligand. The intrasphere mechanism is realized in complexes containing halides, hydroxide ions, and carboxyl groups that can act as bridges between metals. An example is the reaction between pentammine chlorocobalt(III) and hexaaquachrome(II) ions. The process can be roughly divided into three stages: the formation of a heterometallic complex with a bridging chloride ion, electron transfer and decomposition of the bridging complex. The resulting 2+ ion, being labile, instantly turns into an aqua complex, and the inert [(H 2 O) 5 CrCl] 2+ does not interact with water:

If there are no particles in the system that could act as bridges, the process proceeds in the outer sphere:
2+ + 3+ = 3+ + 2+ .
It is especially necessary to highlight the reactions of oxidative addition and reductive elimination, discussed in Chapter 6.
Reactions of coordinated ligands. This group of reactions includes modification processes of ligands coordinated by a metal ion. For example, diketonate complexes, like free diketones, can be nitrated, acylated, or halogenated. The most interesting and unusual example of reactions of coordinated ligands is template synthesis– a unique method of “assembling” a ligand on a metal ion. An example is the synthesis of phthalocyanines from phthalic acid nitrile, which occurs in the presence of copper(II) ions, and the synthesis of a macrocyclic Schiff base from 2-aminobenzaldehyde, which occurs in the presence of nickel(II) ions:

In the absence of metal, the process proceeds along a different path, and the desired product is present in only a small amount in the reaction mixture. The metal ion acts in template synthesis as a matrix (“template”), stabilizing one of the products that are in equilibrium with each other, and shifting the equilibrium towards its formation. For example, in the reaction X + Y ¾® a mixture of products A and B is formed, in which B, which has a lower energy, predominates. In the presence of a metal ion, substance A predominates in the reaction products in the form of a complex with M (Fig. 1.40. Energy diagram of the interaction of X and Y in the absence of a metal ion (left) and in its presence (b)).
Questions and tasks
1. Which of the following compounds has a perovskite structure? BaTiO 3, LiNbO 3, LaCrO 3, FeTiO 3, Na 2 WO 4, CuLa 2 O 4, La 2 MgRuO 6. The table of ionic radii is given in the Appendix. Keep in mind that in complex oxide phases, the B positions may contain cations of two different metals.
2. Using the TCP, determine whether the following spinels will be straight or inverted: ZnFe 2 O 4, CoFe 2 O 4, Co 3 O 4, Mn 3 O 4, CuRh 2 O 4.
3. Thiocyanate ion SCN - has two donor centers - hard and soft. Predict what structure the thiocyanate complexes of calcium and copper(I) will have. Why is it not possible to obtain copper(II) thiocyanate?
4. The spectrum of the Cr 2+ aqua ion (ground state term 5 D) has two bands (Fig. 1.41. Spectrum of the Cr 2+ aqua ion), although among the terms of the nearest excited states there is not one with the same multiplicity. What explains this? What color does this ion have?
5. Using the Δο values below, calculate the ESC for the following complexes in kJ/mol:
(a) 2–, Δο = 15000 cm–1,
(b) 2+, Δο = 13000 cm–1,
(c) 2–, Δο (for 4–)= 21000 cm–1,
Take the pairing energy equal to 19000 cm –1, 1 kJ/mol = 83 cm –1. Calculate their magnetic moments (spin component).
6. Using TCP, explain why the CN – ion reacts with hexaaquanickel(III) ion to form hexacyanoferrate(II), and with hexaaquanickel(II) ion to form tetracyanonickelate(II).
7. Below are the reaction constants for the sequential replacement of water in the copper(II) aqua complex with ammonia: K 1 = 2´10 4 , K 2 = 4´10 3 , K 3 = 1´10 3 , K 4 = 2´10 2 , K5 = 3´10 –1, K6<< 1. Чем объясняется трудность вхождения пятой и шестой молекул аммиака в координационную сферу меди?
8. How does the rigidity of cations change when moving along a 3d row? Is this consistent with the order of change in the stability constants of the complexes (Irving-Williams series, Fig. 1.34).
9. Explain why the hexaquatic iron (III) ion is colorless, and solutions of iron (III) salts are colored.
10. Suggest a mechanism for the reaction 3– + 3– = 4– + 2–, if it is known that the introduction of thiocyanate ion into the solution leads to a change in the reaction rate, and the rate is practically independent of the presence of ammonia. Offer an explanation for these facts.
The concept of changes in the electronic structure of transition metal ions under the action of the electric field of charged particles surrounding them was proposed by Becquerel and further developed by H.A. Bethe and J. Van Vleck at the beginning XX V. These concepts were applied to the description of the electronic structure and properties of complex compounds only in the middle XX century by H. Hartmann and the model was called “crystal field theory” (CFT).
Basic provisions of TCH for transition complexes d metals Fig. 24):
1. - The complex exists and is stable due to the electrostatic interaction of the complexing agent with the ligands.
2. - Ligands are considered without taking into account their electronic structure as point charges or dipoles.
3. - Under the influence of the electric field of the ligands, valence fivefold degenerate ( n -1) d orbitals are split depending on the symmetry of the ligand environment.
4. - Distribution of metal valence electrons among split ( n -1) d orbitals depends on the ratio of the spin-pairing energy and the splitting energy.
Consider, for example, the change in the energy of fivefold degenerate ( n -1) d orbitals of the central metal ion M n+ , located at the center of coordinates, under the influence of the octahedral field of negatively charged ligands [ ML 6] z , located on the coordinate axes (Fig. 25). As a result of the repulsion of the valence electrons of the metal from negatively charged ligands with a uniform distribution of negative charge around the metal (spherically symmetrical electric field), the energy of all five d orbitals will increase by the amount E 0 compared to free M n+ ion. Because the d orbitals have different spatial orientations, then with the concentration of negative charges on ligands located on the coordinate axes, the increase in their energy differs. Energy Boost d z 2 and d x 2- y 2 orbitals directed towards the ligands on the coordinate axes have a greater energy increase dxy, dxz and dyz orbitals directed between coordinate axes.
Energy of fissionfivefold degenerate ( n -1) orbitals into doubly degenerate d x 2- y 2, z 2 orbitals and triply degenerate d xy, xz, yz orbitals are called (Fig. 26) crystal field splitting parameter. Since the energy of the split d orbitals in the octahedral field of the ligands does not change compared to the spherically symmetric electric field, then the increase in the energy of the two d x 2- y 2, z 2 orbitals occurs at 0.6D 0 and a decrease in the energy of three d xy , xz , yz orbitals by 0.4 D 0 .
To indicate the degree of degeneracy and symmetry of metal orbitals split under the influence of the electric field of ligands, special symbols are used. Triple degenerate and symmetric with respect to the center of symmetry and rotation around the coordinate axes d xy , xz , yz t 2 g ", while doubly degenerate and also symmetric with respect to the center of symmetry d x 2- y 2, z 2 orbitals are designated by the symbol " e g " Thus, under the influence of the octahedral electric field of the ligands, fivefold degenerate ( n -1) d the orbitals of the complexing agent are split into triply and doubly degenerate ones of different energies t 2 g and e g orbitals.
A similar consideration of the change in energy of fivefold degenerate ( n -1) d orbitals of a free metal ion in a tetrahedral environment of ligands in [ ML 4 ]z complexes shows (Fig. 27) their splitting also into twofold (e) and threefold ( t ) degenerate orbitals, however, with the opposite energy position. Subscript " g " when designated "e" and " t » orbitals are not indicated since the tetrahedral complex does not have a center of symmetry. A decrease in the number of ligands of a tetrahedral complex compared to an octahedral complex leads to a natural decrease in the crystal field splitting parameter:D T = 4/9 D ABOUT .
Reducing the symmetry of the ligand environment of the metal, for example, tetragonal distortion of octahedral [ ML 6] z complexes associated with the extension of metal-ligand bonds with axial ligands [ ML 4 X 2 ] z and the formation in the limiting case of plane-square [ ML 4 ]z complexes, leads (Fig. 28) to additional splitting of valence ( n -1) d metal orbitals.
Filling of split ( n -1) d metal orbitals occurs in accordance with the Pauli principles and minimum energy. For octahedral complexes with d 1 , d 2 and d 3 electronic configuration of the metal, valence electrons, in accordance with Hund’s rule, populate t 2 g orbitals with parallel spins, leading to t 2 g 1 , t 2 g 2 and t 2 g 3 electronic structure of complexes.
For metals with d 4 electronic configuration, three electrons also populate t 2 g orbitals with parallel spins. The population of the fourth electron depends on the energy costs for the value of the spin pairing energy (E sp.-sp.) during the population t 2 g orbitals with antiparallel spin and violation of Hund's rule, or overcoming the energy of splitting by the crystal fieldD o upon check-in e g orbitals with parallel spin in accordance with Hund's rule. In the first case, a complex is formed with t 2 g 4 electronic structure and reduced spin multiplicity compared to free metal 2 S +1 = 3 (S - total spin), called low-spin. When Hund's rule is fulfilled and the fourth electron is populated on e g orbitals, a complex is formed with t 2 g 3 e g 1 electronic structure and free metal-like spin multiplet 2 S +1 = 5. Such complexes are called high-spin.
Similarly, when distributing valence d5, d6 and d7 metal electrons t 2 g and e g orbitals of octadric complexes depending on the ratio E sp.-sp. AndD O The formation of two types of complexes is possible:
At E sp.-sp. > D O high-spin complexes with the electronic structure of the metal are formed t 2 g 3 e g 2 , t 2 g 4 e g 2 , t 2 g 5 e g 2 according to Hund's rule and free metal-like spin multiplicity - 2 S +1 = 6, 5, 4;
E sp.-sp.< D O low-spin complexes with the electronic structure of the metal are formed t 2 g 5 e g 0 , t 2 g 6 e g 0 , t 2 g 6 e g 1 and lower spin multiplicity compared to free metal 2 S +1 = 2, 1, 2.
Metal complexes with d 8, d 9 and d 10 electronic configuration are characterized by one type of electron distribution - t 2 g 6 e g 2 , t 2 g 6 e g 3 , t 2 g 6 e g 4 with spin multiplicity similar to free metal: 2 S +1 = 3, 2 and 0.
So the parameterD, characterizing the splitting ( n -1) d metal orbitals under the influence of the electric field of the ligands is one of the main characteristics of changes in the properties of complexes compared to a free metal ion. It is the parameter valueDdetermines for a number of electronic configurations of the metal determines the possibility of the formation of high- or low-spin complexes with different distributions of electrons over split orbitals and different properties.
The value of the crystal field splitting parameterDdepends on the nature of the metal of the complexing agent, the ligands surrounding it and their spatial position around the complexing agent:
1. Ligands in order of increasing parameterDfor complexes of the same metal and similar geometric structure are located in the so-called spectrochemical series: I -< Br - < Cl - < F - < OH - < C 2 O 4 2- ~ H 2 O < NCS - < NH 3 ~ En < NO 2 - < CN - < CO . At the beginning of the row there are “weak field” ligands - halide ions, hydroxide and oxalate ions, water, which form predominantly high-spin complexes. The ligands on the right side of the series: carbon monoxide, cyanide and nitrite ions are called “high field” ligands and are typically characterized by the formation of low-spin complexes. For ligands in the middle of the series - thiocyanate ion, ammonia, ethylenediamine, depending on the nature of the metal, high- or low-spin complexes are formed.
2. Increasing the efficiency of the electric field of ligands on d metal orbitals with increasing their size in row 3 d<< 4 d < 5 d , as well as an increase in the degree of oxidation of the metal leads to an increase in the parameterD in the series: Mn(II)< Ni (II ) < Co (II ) < Fe (II ) < V (II ) < Fe (III ) < Co (III ) < Mn (IV ) < Mo (III ) < Rh (III ) < Ru (III ) < Pd (IV ) < Ir (III ) < Pt (IV ).
3. Parameter Dfor tetrahedral complexes is only 4/9 of the parameterDoctahedral complexes.
“Heavy” complexes 4 d and 5 d metals, almost regardless of the nature of the ligands, form predominantly low-spin complexes, while the formation of low- or high-spin complexes is “light” 3 d metals is mainly determined by the strength of the ligand field.
In contrast to the MMS, the crystal field theory to justify the difference in the magnetic properties of complexes of the same metal ion with different ligand environments, for example, diamagnetic [ Fe(CN ) 6 ] 4- and paramagnetic [ Fe(H2O ) 6 ] 2+ does not use the hypothesis of their intraorbital ( d 2 sp 3 hybridization) and outer-orbital ( sp 3 d 2 hybridization) structure. The difference in magnetic properties is determined by the low- and high-spin nature of the distribution of 6-valent electrons Fe(II ) by split t 2 g and e g orbitals (Fig. 29). Being strong and weak field ligands, cyanide ions and water molecules form Fe(II ) low- and high-spin complexes with t 2 g 6 e g 0 and t 2 g 4 e g 2 distribution of electrons, which determines diamagnetism [ Fe(CN ) 6 ] 4- and paramagnetism [ Fe(H2O ) 6 ] 2+ complexes.
Splitting of fivefold degenerate ( n -1) d metal orbitals in complexes and parameter changesDdepending on the nature of the ligands, it determines the characteristic color of the complexes both in the solid state and in solutions. When the complex absorbs electromagnetic radiation in the visible region of the spectrum (400-750) nm, the energy of the quanta of which is E equal to the value D, electron transfer occurs from t 2 g on e g orbitals. It is the unabsorbed electromagnetic radiation of the visible region of the spectrum that determines the color of the complex in accordance with the “Newton’s color circle” (Fig. 30), showing the primary and secondary colors of visible radiation.
Aquacomplex titanium( III) [Ti (H 2 O) 6] 3+ c t 2 g 1 e g 0 electronic distribution as a result of photoexcitation, corresponding to the transition of the electron to higher energy e g orbitals:
3+ (t 2g 1 e g 0) + hn= * 3+ (t 2g 0 e g 1)
absorbs light quanta in the yellow region of the spectrum, which leads to its violet color. A change in the ligand environment of the metal ion in accordance with the position of the ligand in the spectrochemical series leads to a change in the parameterDand, as a consequence of this, to a change in the energy and wavelength of quanta absorbed by the complex and to the characteristic color of the complex - for example, in the series [ CuCl 4 ] 2- , [ Cu (H 2 O ) 4 ] 2+ , [ Cu (NH 3 ) 4 ] 2+ the color of the complexes changes from green to blue and violet.
Along with the crystal field splitting energyD, also plays an important role in TCH crystal field stabilization energy(ESKP) - gain in energy when distributing electrons among those split in the complex ( n -1) d metal orbitals compared to the energy of fivefold degenerate ( n -1) d metal orbitals in an equivalent spherical electric field (Fig. 31, 32).
ESCP of octadral and tetrahedral complexes.|
Mn+ |
Octahedral complexes |
Tetrahedral complexes |
|
|
Low spin |
High spin |
High spin |
|
|
0.4 D o |
0.6 D T |
||
|
0.8 D o |
1.2 D T |
||
|
1.2 D o |
0.8 D T |
||
|
d 4 |
1.6 D o |
0.6 D o |
0.4 D T |
|
d 5 |
2.0 D o |
0 D o |
0 D T |
|
d 6 |
2.4 D o |
0.4 D o |
0.6 D T |
|
d 7 |
1.8 D o |
0.8 D o |
1.2 D T |
|
d 8 |
1.2 D o |
0.8 D T |
|
|
d 9 |
0.6 D o |
0.4 D T |
|
|
d 10 |
0 D o |
||
An estimate of the EXP value of the complex is obtained on the basis of splitting diagrams ( n -1) d metal orbitals in the electric field of ligands, showing a decrease or increase in the energy of the system compared to a spherical electric field when electrons populate split ( n -1) d orbitals. For octahedral [ ML 6] z complexes (Fig. 32) population of each electron t 2 g orbitals leads to a gain in system energy by 0.4D oh, check-in e g requires energy expenditure 0.6D O . For tetrahedral [ ML 4 ]z complexes with opposite energy positions e and t metal orbitals - occupation of each electron by split e and t orbitals is accompanied by a decrease and increase in the energy of the system by 0.6D t and 0.4 D T .
Being a reflection of the thermodynamic stability of the complexes, estimates of their ESCR values are consistent with experimental data on changes in the energy of the crystal lattice for high-spin hexafluoride complexes 3 d metals (Fig. 33).
ESC values allow us to determine the most preferred coordination isomer (Fig. 34), for example [ Cu (NH 3 ) 6 ][ NiCl 4 ] or [ Ni (NH 3 ) 6 ][ CuCl 4 ]. To do this, calculate the difference in ESC for the complex cation and anion of the isomers. ESCR value [ Cu (NH 3 ) 6 ] 2+ and [NiCl 4 ] 2- is 0.6 D o and 0.8 D T respectively. Considering thatD t = 4/9 D o , the difference between the ESCP values [ Cu (NH 3 ) 6 ] 2+ and [NiCl 4 ] 2- will be 19/45D o . Similarly, the values of ESKP [ Ni (NH 3 ) 6 ] 2+ and [CuCl 4 ] 2- is 1.2 D o and 0.4 D T , and the difference between them is 28/45D o . Big difference ESCP complex cation [ Ni (NH 3 ) 6 ] 2+ and the anion [CuCl 4 ] 2- compared to [ Cu (NH 3 ) 6 ] 2+ and [NiCl 4 ] 2- shows a more preferable formation of the isomer of composition [ Ni (NH 3 ) 6 ][ CuCl 4 ].
Along with the magnetic and optical properties of the influence of the electronic structure of the metal on the thermodynamic stability of the complexes, TKP predicts a distortion of the geometric structure of the complexes with an uneven distribution of electrons over split ( n -1) d metal orbitals (Fig. 35). In contrast to the regular octahedral structure [ Co (CN) 6 ] 3- s t 2 g 6 e g 0 electronic distribution, tetragonal distortion of a similar complex [ Cu (CN) 6 ] 4- s t 2 g 6 e g 3 electronic distribution containing 3 electrons on 2-fold degenerate e g orbitals, leads to the effective transformation of the octahedral into a square-planar complex:
4- = 2- + 2CN - .
All of the above shows that the relative simplicity and broad capabilities of TCT for explaining and predicting the physicochemical properties of complexes determine the great popularity of this model for describing chemical bonds in complex compounds. At the same time, focusing on changes in the electronic structure of the metal during complex formation, TCP does not take into account the electronic structure of the ligands, considering them as point negative charges or dipoles. This leads to a number of limitations of TCP when describing the electronic structure of complexes. For example, within the framework of TCP it is difficult to explain the position of a number of ligands and metals in spectrochemical series, which is associated with a certain degree of covalence and the possibility of the formation of multiple metal-ligand bonds. These limitations are eliminated when considering the electronic structure of complex compounds using the more complex and less visual method of molecular orbitals.
Valence bond theory was the first of the quantum mechanical theories used to approximately explain the nature of chemical bonds in complex compounds. Its application was based on the idea of donor-acceptor mechanism formation of covalent bonds between the ligand and the complexing agent. Ligand counts donor particle, capable of transferring a pair of electrons acceptor – complexing agent, which provides free quantum cells (atomic orbitals) of its energy levels for the formation of bonds.
For the formation of covalent bonds between the complexing agent and ligands, it is necessary that the vacant s-, p- or d-atomic orbitals of the complexing agent have undergone hybridization a certain type. Hybrid orbitals occupy a certain position in space, and their number corresponds to coordination number complexing agent.
This often happens combining unpaired electrons complexing agent into pairs, which allows the release of a certain number of quantum cells - atomic orbitals, which then participate in hybridization and the formation of chemical bonds.
The lone pairs of electrons of the ligands interact with the hybrid orbitals of the complexing agent, and overlap corresponding orbitals of the complexing agent and ligand with the appearance of increased electron density in the internuclear space. The electron pairs of the complexing agent, in turn, interact with the vacant atomic orbitals of the ligand, strengthening the connection through the dative mechanism. Thus, chemical bonding in complex compounds is common covalent sufficient connection durable And energetically favorable.
Electron pairs located in the hybrid orbitals of the complexing agent tend to occupy a position in space in which their mutual repulsion is minimal. This leads to structure complex ions and molecules appears to be in a certain dependence on type of hybridization.
Let us consider the formation of some complexes from the standpoint of the theory of valence bonds. First of all, we note that the valence orbitals of the atoms of the complexing agents are close in energy:
E (n- 1)d » E ns » E n.p. » E nd
|
Hybridization type |
Geometry of the complex |
||
|
linear |
-
|
||
|
triangular |
- |
||
|
tetrahedron |
2-
|
||
|
2-
|
|||
|
sp 3 d(z 2) |
trigonal bipyramid |
||
|
sp 3 d(x 2 - y 2) |
square pyramid |
3-
|
|
|
sp 3 d 2 , |
3+ |
||
|
sp 3 d 3 |
pentagonal bipyramid |
4-
|
For example, the 2+ cation includes the complexing agent zinc(II). The electron shell of this conventional ion has the formula 3 d 10 4s 0 4p 0 and can be conventionally depicted as follows:
Vacant 4 s- and 4 p-orbitals of the zinc(II) atom form four sp 3-hybrid orbitals oriented towards the vertices of the tetrahedron.
Each ammonia molecule has a lone pair of electrons on the nitrogen atom. The orbitals of nitrogen atoms containing lone pairs of electrons overlap with sp 3-hybrid orbitals of zinc(II), forming a tetrahedral complex cation of tetraammine zinc(II) 2+: 
Since the 2+ ion has no unpaired electrons, it exhibits diamagnetic properties.
Tetrachloromanganate(II) ion 2- contains five unpaired electrons per 3 d-orbitals and vacant 4 s- and 4 p-orbitals. Vacant orbitals form sp 3-hybrid orbitals that overlap with p-atomic orbitals of chloride ions: 
The tetrahedral ion 2- thus obtained is paramagnetic, since it contains five unpaired electrons.
Using a conventional prediction algorithm type of hybridization of atomic orbitals within the framework of the valence bond method, one can determine geometry of complexes of different composition. To do this, first of all, it is necessary to write the electronic formula for the valence level and construct a diagram of the distribution of electrons over quantum cells. For example, for a neutral nickel atom:

Transition 4 s-electrons for 3 d-sublevel transforms paramagnetic Ni atom 0 in diamagnetic particle Ni*: 
The resulting vacant orbitals undergo hybridization, forming a tetrahedral configuration. Built like that tetrahedral diamagnetic tetracarbonylnickel complex (CN = 4), which is characterized by significant stability.
If the complexing agent is nickel(II) with electron configuration 3 d 8 4s 0 4p 0, then the need to move electrons from 4 s-sublevel before hybridization disappears, since there is a sufficient number of vacant orbitals to realize the coordination number 4: 
This structure has an unstable paramagnetic tetrabromoniccolate(II)-ion 2- complex. However, when combining two electrons 3 d-sublevel into a pair and the transformation of one of the quantum cells of this sublevel into a vacant one changes both the type of hybridization and the characteristics of the resulting complex: 
Hybridization type dsp 2 and the planar square shape of the complex are realized upon the formation of a stable diamagnetic complex tetracyanoniccolate(II)-ion 2- (CN = 4): 
If the synthesis of the cyanide complex is carried out under conditions of excess ligand, a coordination number of 5 can be realized: 
Stable diamagnetic the pentacyanoniccolate(II)-ion 3- complex has the shape of a square pyramid: 
Octahedral nickel(II) 2+ complex, although paramagnetic, but quite stable. His education is due sp 3 d 2 -hybridization of nickel atomic orbitals: 
If atomic orbitals of the outer d-sublevel, complex, as a rule, to a large extent paramagnetic and is called outer-orbital or high-spin. The structure of such complexes may correspond to the type of hybridization, for example, sp 3 d 2 .
Such complexes, during the formation of which hybridization takes place with the participation of atomic orbitals of the outer d-sublevels are called intra-orbital or low-spin and, as a rule, diamagnetic or weakly paramagnetic(all or almost all electrons of the complexing agent are paired, and the type of hybridization, e.g. d 2 sp 3 or dsp 2).
When examining iron(II) complexes, both outer orbital and intraorbital complexes are found.


The diagram below shows how they are formed paramagnetic high-spin hexafluoroferrate(II)-ion 4- and diamagnetic low-spin hexacyanoferrate(II) ion 4-.
The theory of valence bonds itself does not answer the question of what type of complex is formed in each specific case, since this method does not take into account the influence of the nature of the ligand. Therefore, the valence bond method must necessarily be supplemented with data on the magnetic properties of the complex or information on the influence of the ligand on the nature of the formed complex.
.Crystal field theory replaced the theory of valence bonds in the 40s of the 20th century. In its pure form, it is not currently used, since it cannot explain the formation of covalent bonds in complex compounds and does not take into account the true state of the ligands (for example, their actual sizes) even in the case of interactions close to purely electrostatic.
Already in the mid-50s, the simplified crystal field theory was replaced by an improved ligand field theory, taking into account the covalent nature of the chemical bonds between the complexing agent and the ligand.
However, the most general approach to explaining the formation of complex compounds is given by molecular orbital theory(MO), which currently prevails over all others. The molecular orbital method provides for both purely electrostatic interaction in the absence of overlapping atomic orbitals, and the entire set of intermediate degrees of overlap.
Let's look at the basic concepts crystal field theory, which, like the theory of valence bonds, still retains its importance for the qualitative description of chemical bonds in complex compounds due to its great simplicity and clarity.
In crystal field theory, the chemical bond between the complexing agent and the ligand is considered electrostatic. According to this theory, the ligands are located around the complexing agent at the vertices of regular polyhedra ( polyhedra) as point charges. The theory does not take into account the actual volume of the ligand.
Ligands, like point charges, create around the complexing agent electrostatic field(“crystal field”, if we consider a crystal of a complex compound, or ligand field), in which the energy levels of the complexing agent and, above all, d-sublevels are splitting, and their energy changes. The nature of the splitting, the energy of new energy levels depends on symmetry arrangement of ligands (octahedral, tetrahedral or other crystal field). When molecules H 2 O, NH 3 , CO and others are coordinated as ligands, they are considered as dipoles, oriented with a negative charge towards the complexing agent.
Let us consider the case of an octahedral arrangement of ligands (for example, 3- or 3+). In the center of the octahedron there is a complexing atom M(+n) with electrons on d-atomic orbitals, and at its vertices there are ligands in the form of point negative charges (for example, F - ions or polar molecules like NH 3). In a conventional ion M(+n) not associated with ligands, the energies of all five d-AO are the same (i.e. atomic orbitals degenerate).
However, in the octahedral field of ligands d-AOs of the complexing agent fall into unequal position. Atomic orbitals d(z 2) and d(x 2 -
y 2), elongated along the coordinate axes, come closest to the ligands. Between these orbitals and the ligands located at the vertices of the octahedron, significant differences arise repulsive forces, leading to an increase in orbital energy. In other words, these atomic orbitals are subject to maximum exposure to the ligand field. A strongly compressed spring can serve as a physical model of such interaction. 

Other three d-AO – d(xy), d(xz) And d(yz), located between the coordinate axes and between the ligands, are at a greater distance from them. The interaction of such d-AO with ligands is minimal, and therefore energy d(xy), d(xz) And d(yz)-AO decreases compared to the original one. 

Thus, fivefold degenerate d-AO complexing agent, getting into octahedral ligand field, exposed splitting into two groups of new orbitals – triply degenerate orbitals with lower energy, d(xy), d(xz) And d(yz), And doubly degenerate orbitals with higher energy d(z 2) and d(x 2 -
y 2). These new groups d-orbitals with lower And higher energy denote d e and d g: 
Energy difference two new sublevels d e and d g got the name splitting parameter D0:
E 2 – E 1 = D0
Location of two new energy sublevels d e and d g relative to the original ( d-AO) on the energy diagram asymmetrical:
(E 2 – E 0) > (E 0 – E 1).
Quantum mechanical theory requires that when new energy levels are completely populated with electrons, the total energy remains unchanged, i.e. she should stay equal to E 0 .
In other words, the equality must be satisfied
4(E 2 – E 0) = 6(E 0 – E 1),
where 4 and 6 – maximum number of electrons per d g - and d e -AO. From this equality it follows that
(E 2 – E 0) / (E 0 – E 1) = 3/2 and
(E 2 – E 1) / (E 0 – E 1 >) = 5/2, or
D0/( E 0 – E 1) = 5/2, whence ( E 0 – E 1) = 2/5 ´ D 0 >. Placing each electron out of the maximum six possible on d e-orbitals causes decrease (winnings) energy by 2/5 D 0 . On the contrary, the placement of each electron out of four possible on d g orbitals cause increase (cost) energy by 3/5 D 0 . If populated with electrons d e - and d g -orbitals completely, then no winning energy will not(just as it won't additional energy consumption): 4 ´ 3/5 ´ D 0 - 6 ´ 2/5 ´ D 0 = 0. But if the original d-AO is populated only partially and contains from 1 to 6 electrons, and these electrons are placed only on d e -AO, then we get significant energy gain. The specificity of each ligand affects the field that this ligand creates - strong or weak. How stronger field ligands than more meaning splitting parameter D0. The study of the splitting parameter is usually based on spectroscopic research. Wavelengths absorption bands complexes l in the crystalline state or in solution, due to the transition of electrons from d e - on d g-AO, associated with splitting parameter D 0 as follows: n = 1/l; D where is Planck's constant h equal to 6.626 ´ 10 - 34 J. s; Splitting parameter, in addition to the type of ligand, depends on the degree of oxidation And nature complexing agent. At increasing nuclear charge of the complex-forming atom D 0 also increases. Hexaamminiridium(III) 3+, hexaamminiridium(III) 3+, and hexaamminiridium(III) 3+ cations ( Z= 27, 45 and 77) are characterized by splitting parameters equal to 22900, 34100 and 41000 cm -1. The dependence of D0 on the nature of the ligands is more diverse. As a result of the study of numerous complex compounds, it was found that in terms of their ability to increase the cleavage parameter of complexing metals located in their usual oxidation states, the most common ligands can be arranged in the following spectrochemical series, along which the value of D 0 monotonically increases: Thus, the strongest electrostatic field around the complexing agent and the strongest cleavage d-AO is caused by NO 2 - ligands, CN -
and CO. Let us consider the distribution of electrons over d e - and d g-orbitals in the octahedral field of ligands. Check-in d e - and d g-orbitals occurs in full accordance with Hund's rule And Pauli principle. In this case, regardless of the value of the splitting parameter, the first three electrons are occupied by quantum cells d e-sublevel: If the number of electrons per d- there are more than three sublevels of the complexing agent; there are two possibilities for placing them on split sublevels. At a low value of the splitting parameter (weak field of the ligands), electrons overcome the energy barrier separating d e - and d g-orbitals; the fourth and then the fifth electrons populate quantum cells d g-sublevel. With a strong ligand field and a high D0 value, the population is populated by the fourth and fifth electrons d g-sublevel excluded; filling in progress d e-orbitals. At weak field ligands populating quantum cells with 4 or 5 electrons parallel spins, so the resulting complex turns out to be strongly paramagnetic. In a strong ligand field one and then two electron pairs are formed on d e -sublevel, so paramagnetism the complex turns out to be much weaker. The sixth, seventh and eighth electrons in the case of a weak field end up back on d e -sublevel, complementing the configurations to electron pairs (one in the case d 6, two – d 7 and three - d 8): In the case of a strong ligand field, the sixth electron populates d e -AO, leading to diamagnetism complex, after which the seventh and eighth electrons go to d g-sublevel: Obviously, with an eight-electron configuration differences in structure between complexes with ligands weak And strong fields disappear. The occupation of orbitals by the ninth and tenth electron also does not differ for complexes of both types: Let us return to the consideration of the electronic structure of the octahedral complex ions 3+ and 3-. According to the location in spectrochemical series, ammonia NH 3 is one of the ligands strong field
, and fluoride ion F - – weak field
. Consequently, the occupation of atomic orbitals by electrons in these complexes will occur according to the following scheme: In the 3- anion, the F - ligands create a weak crystal field (D 0 = 13000 cm - 1), and all the electrons of the original 3 d 6 -JSC are located on d e - and d g orbitals without any pairing. A complex ion is high-spin and contains four unpaired electrons, so it paramagnetic. In the 3+ ion, NH 3 ligands create a strong crystal field (D 0 = 22900 cm - 1), all 3 d 6 -electrons are placed on a more energetically favorable d e-orbitals. Transfer of electrons from d e - on d g-orbitals impossible because too high energy barrier. Therefore, this complex cation is low spin, it does not contain unpaired electrons and diamagnetic. In a similar way, schemes for the distribution of electrons over orbitals in an octahedral field for 2+ and 4- ions can be presented: H 2 O ligands create a weak field; exchange of electrons between d e - and d g-orbitals does not cause any difficulties and therefore the number of unpaired electrons in the complex ion is the same as in the conventional Fe + II ion. The resulting aqua complex is high-spin, paramagnetic. Many complex compounds in the crystalline state and aqueous solution are brightly colored. Thus, an aqueous solution containing 2+ cations is colored intensely blue, 3+ cations give the solution a purple color, and 2+ cations give it a red color. The crystal field theory makes it possible to explain the appearance of one color or another in complex compounds. If light is passed through a solution or crystalline sample of a substance visible part of the spectrum, then, in principle, three options for the physical behavior of the sample are possible: no light absorption any wavelength (substance sample colorless, although it may have absorption bands in the ultraviolet region of the spectrum); complete light absorption over the entire wavelength range (the sample will appear black); finally, light absorption only certain wavelength(then the sample will have color complementary to absorbed narrow part of the spectrum). Thus, the color of the solution or crystals is determined frequency of absorption bands visible light: The absorption of light quanta by complexes (for example, those with an octahedral structure) is explained by the interaction of light with electrons located on d e -sublevel, accompanied by their transition to vacant orbitals d g-sublevel. For example, when passing light through an aqueous solution containing hexaaquatitanium(III) 3+ cations, a light absorption band is detected in the yellow-green region of the spectrum (20300 cm - 1, l » 500 nm). This is due to the transition of the single electron of the complexing agent from d e-AO on d g-sublevel: Therefore, a solution containing 3+ acquires a violet color (in addition to the absorbed yellow-green). A solution of vanadium salt Cl 3 is green. This is also due to the corresponding transitions of electrons when they absorb part of the energy of the light beam. In the ground state, with the electronic configuration of vanadium(III) 3 d 2, two unpaired electrons are occupied d e-sublevel: There is only two options for the transition of two electrons on d g -sublevel: either both electrons are occupied d g -AO, or only one of them. Any other electron transitions associated with a decrease in the total spin are prohibited. If the complexing agent has an electronic configuration d 0 or d 10 then electron transitions With d e - on d g -sublevel or vice versa impossible either because absence of electrons, either because absence of vacant orbitals. Therefore, solutions of complexes with such complexing agents as Sc(III), Cu(I), Zn(II), Cd(II), etc., do not absorb energy in the visible part of the spectrum and appear colorless: The selectivity of light absorption depends not only on complexing agent And its oxidation state, but also from type of ligands. When replacing ligands on the left side of the spectrochemical series in a complex compound with ligands that create strong electrostatic field observed increase the fraction of energy absorbed by electrons from transmitted light and, as a consequence, decrease wavelength of the corresponding absorption band. Thus, an aqueous solution containing tetraaquacopper(II) 2+ cations is blue, and a solution of tetraamminecopper(II) 2+ sulfate is intensely blue. ________________________ Repeat: >>> Applications
Energy gain due to priority settlement electrons d e-atomic orbitals are called energy of stabilization of the complex by the ligand field.
speed of light With
= 3 ´ 10 10 cm/s.
Unit D 0 is the same as that of the wave number n: cm - 1, which approximately corresponds to 12 J/mol.
In complex compounds that include complexing agents of the same period and in the same oxidation state, with the same ligands, the cleavage parameter is approximately the same. With increasing degree of oxidation of the complexing agent, the value of D 0 increases. Thus, for aqua complexes 2+ and 2+, the value of the splitting parameter is 7800 and 10400 cm - 1, and for 3+ and 3+ - 13700 and 21000 cm - 1, respectively.
I-Br - Cl - » NCS - NO 3 - F -OH - H2O » H - NH 3 NO 2 -CN - "NO" CO. 









Conversely, CN - ligands cause significant cleavage d-AO, amounting to 33000 cm - 1. This means that there is a strong tendency to allocate all electrons on d e-orbitals. Energy Gain, obtained with such a population of orbitals, is much greater than the energy costs due to electron pairing.

The indicated transitions of electrons that have received excess energy correspond to absorption band about 400 nm in the absorption spectrum of a solution of hexaaquavanadium(III) chloride. Absorption of the purple-violet region of the spectrum gives an additional color to the solution - bright green.

And John Van Vleck to describe the lower states of transition metal cations surrounded by ligands - both anions and neutral molecules. Crystal field theory was further combined [and refined] with (delocalized) molecular orbital theory into a more general theory that takes into account the partial covalency of the metal-ligand bond in coordination compounds.
Crystal field theory allows one to predict or interpret the optical absorption spectra and electron paramagnetic resonance spectra of crystals and complex compounds, as well as the enthalpies of hydration and stability in solutions of transition metal complexes.
Review of Crystal Field Theory[ | ]
According to TCP, the interaction between a transition metal and ligands arises from the attraction between the positively charged metal cation and the negative charge of electrons in the nonbonding orbitals of the ligand. The theory considers the change in energy of five degenerate d-orbitals surrounded by point charges of ligands. As the ligand approaches the metal ion, the ligand's electrons become closer to some d-orbitals than others, causing a loss of degeneracy. Electrons d-orbitals and ligands repel each other as charges with the same sign. Thus, the energy of those d-electrons that are closer to the ligands become higher than those that are further away, which leads to a splitting of energy levels d-orbitals.
The following factors influence splitting:
- Nature of the metal ion.
- The degree of oxidation of the metal. The higher the oxidation state, the higher the cleavage energy.
- Arrangement of ligands around a metal ion.
- The nature of the ligands surrounding the metal ion. The stronger the effect of the ligands, the greater the difference between high and low energy levels.
The most common type of ligand coordination is octahedral, in which six ligands create a crystal field of octahedral symmetry around the metal ion. In the octahedral environment of a metal ion with one electron in the outer shell, the d-orbitals are divided into two groups with a difference in energy levels Δ oct ( fission energy), while the energy of the orbitals dxy, dxz And d yz will be lower than d z 2 and d x 2 -y 2, since the orbitals of the first group are located further from the ligands and experience less repulsion. The three low energy orbitals are designated as t 2g, and two with high - like e g.
The next most common are tetrahedral complexes in which four ligands form a tetrahedron around a metal ion. In this case d-orbitals are also divided into two groups with a difference in energy levels Δ tetr. Unlike octahedral coordination, the orbitals will have low energy d z 2 and d x 2 -y 2, and high - d xy , d xz And d yz. In addition, since the electrons of the ligands are not directly in the direction d-orbitals, the splitting energy will be lower than with octahedral coordination. Using TCP you can also describe plano-square and other geometries of complexes.
The difference in energy levels Δ between two or more groups of orbitals also depends on the nature of the ligands. Some ligands cause less cleavage than others, the reasons for which are explained. Spectrochemical series- an experimentally obtained list of ligands, ordered in ascending order Δ:
The oxidation state of the metal also affects Δ. A metal with a higher oxidation state attracts ligands closer due to a larger charge difference. Ligands closer to the metal ion cause more cleavage.
Low- and high-spin complexes[ | ]
Ligands that cause major cleavage d-levels, such as CN− and CO, are called ligands strong field. In complexes with such ligands, it is unfavorable for electrons to occupy high-energy orbitals. Consequently, low energy orbitals are completely filled before high energy orbitals begin to fill. Such complexes are called low-spin. For example, NO 2 − is a high-field ligand that produces large splitting. All 5 d-electrons of the octahedral ion 3− will be located at the lower level t 2g .
In contrast, ligands that cause little cleavage, such as I− and Br−, are called ligands weak field. In this case, it is easier to place electrons in high energy orbitals than to place two electrons in the same low energy orbital, because two electrons in the same orbital repel each other, and the energy cost of placing a second electron in the orbital is higher than Δ. Thus, before paired electrons appear, in each of the five d-orbitals must be placed one electron at a time in accordance with Hund's rule. Such complexes are called high-spin. For example, Br− is a weak-field ligand causing little splitting. All 5 d-orbitals of the 3− ion, which also has 5 d-electrons will be occupied by one electron.
The splitting energy for tetrahedral complexes Δ tetr is approximately equal to 4/9Δ oct (for the same metal and ligands). As a result, the difference in energy levels d-orbitals are usually below the electron pairing energy, and tetrahedral complexes are usually high-spin.
Distribution diagrams d-electrons make it possible to predict the magnetic properties of coordination compounds. Complexes with unpaired electrons are paramagnetic and are attracted by a magnetic field, while complexes without are diamagnetic and weakly repel.
Crystal field stabilization energy[ | ]
Crystal field stabilization energy (CFE) is the energy of the electronic configuration of a transition metal ion relative to the average energy of the orbitals. Stabilization occurs due to the fact that in the ligand field the energy level of some orbitals is lower than in a hypothetical spherical field, in which all five d-orbitals have the same repulsive force, and all d-orbitals are degenerate. For example, in the octahedral case the level t 2g lower than the average level in a spherical field. Consequently, if these orbitals contain electrons, then the metal ion is more stable in the ligand field relative to the spherical field. On the contrary, the energy level of the orbitals e g above average, and the electrons contained in them reduce stabilization.
Energy of stabilization by the octahedral field
There are three orbitals in an octahedral field t 2g stabilized relative to the average energy level by 2/5 Δ octave, and the two orbitals e g destabilized by 3/5 Δ oct. Above were examples of two electronic configurations d 5 . In the first example, there is a low-spin complex 3− with five electrons in t 2g. Its ESP is 5 × 2 / 5 Δ oct = 2Δ oct. In the second example, a high-spin complex 3− with ESKP (3 × 2 / 5 Δ oct) − (2 × 3 / 5 Δ oct) = 0. In this case, the stabilizing effect of electrons in low-level orbitals is neutralized by the destabilizing effect of electrons in high-level orbitals.
Diagrams of d-level splitting by a crystal field[ | ]
| octahedral | pentagonal-bipyramidal | square-antiprismatic |
|---|---|---|
Crystal field theory replaced the theory of valence bonds in the 40s of the 20th century. In its pure form, it is not currently used, since it cannot explain the formation of covalent bonds in complex compounds and does not take into account the true state of the ligands (for example, their actual sizes) even in the case of interactions close to purely electrostatic.
Already in the mid-50s, the simplified crystal field theory was replaced by an improved ligand field theory, taking into account the covalent nature of the chemical bonds between the complexing agent and the ligand.
However, the most general approach to explaining the formation of complex compounds is given by molecular orbital theory(MO), which currently prevails over all others. The molecular orbital method provides for both purely electrostatic interaction in the absence of overlapping atomic orbitals, and the entire set of intermediate degrees of overlap.
Let's look at the basic concepts crystal field theory, which, like the theory of valence bonds, still retains its importance for the qualitative description of chemical bonds in complex compounds due to its great simplicity and clarity.
In crystal field theory, the chemical bond between the complexing agent and the ligand is considered electrostatic. According to this theory, the ligands are located around the complexing agent at the vertices of regular polyhedra ( polyhedra) as point charges. The theory does not take into account the actual volume of the ligand.
Ligands, like point charges, create around the complexing agent electrostatic field(“crystal field”, if we consider a crystal of a complex compound, or ligand field), in which the energy levels of the complexing agent and, above all, d-sublevels are splitting, and their energy changes. The nature of the splitting, the energy of new energy levels depends on symmetry arrangement of ligands (octahedral, tetrahedral or other crystal field). When molecules H 2 O, NH 3 , CO and others are coordinated as ligands, they are considered as dipoles, oriented with a negative charge towards the complexing agent.
Consider the case of an octahedral arrangement of ligands (for example, -3 or 3+). In the center of the octahedron there is a complexing ion M(+ n) with electrons on d-atomic orbitals, and at its vertices there are ligands in the form of point negative charges (for example, F - ions or polar molecules like NH 3). In a conventional ion M(+ n), not associated with ligands, the energies of all five d-AO are the same (i.e. atomic orbitals degenerate).
However, in the octahedral field of ligands d-AOs of the complexing agent fall into unequal position. Atomic orbitals d(z 2) and d(x 2 -y 2), elongated along the coordinate axes, come closest to the ligands. Between these orbitals and the ligands located at the vertices of the octahedron, significant differences arise repulsive forces, leading to an increase in orbital energy. In other words, these atomic orbitals are subject to maximum exposure to the ligand field. A strongly compressed spring can serve as a physical model of such interaction.
Other three d-AO – d(xy), d(xz) And d(yz), located between the coordinate axes and between the ligands, are at a greater distance from them. The interaction of such d-AO with ligands is minimal, and therefore energy d(xy), d(xz) And d(yz)-AO decreases compared to the original one.
Thus, fivefold degenerate d-AO complexing agent, getting into octahedral ligand field, exposed splitting into two groups of new orbitals – triply degenerate orbitals with lower energy, d(xy), d(xz) And d(yz), And doubly degenerate orbitals with higher energy d(z 2) and d(x 2 -y 2). These new groups d-orbitals with lower And higher energy denote dε and dγ:
d(z 2) and d(x 2 -y 2)
d(xy), d(xz),d(yz)
Energy difference two new sublevels dε and dγ was named splitting parameter Δ 0:
E 2 – E 1 = Δ 0 ≈ 0
Location of two new energy sublevels dε and dγ relative to the original ( d-AO) on the energy diagram asymmetrical:
(E 2 – E 0) > (E 0 – E 1).
Quantum mechanical theory requires that when new energy levels are completely populated with electrons, the total energy remains unchanged, i.e. she should stay equal to E 0 .
In other words, the equality must be satisfied
4(E 2 – E 0) = 6(E 0 – E 1),
where 4 and 6 – maximum number of electrons per dγ- and dε-AO. From this equality it follows that
(E 2 – E 0) / (E 0 – E 1) = 3/2 and
(E 2 – E 1) / (E 0 – E 1) = 5/2, or
Δ 0 / ( E 0 – E 1) = 5/2, whence ( E 0 – E 1) = 2/5Δ 0 .
Placing each electron out of the maximum six possible on dε orbitals cause decrease (winnings) energy by 2/5 Δ 0 .
On the contrary, the placement of each electron out of four possible on dγ orbitals cause increase (cost) energy by 3/5 Δ 0 .
If populated with electrons dε- and dγ-orbitals completely, then no winning energy will not(just as it won't additional energy consumption).
But if the original d-AO is populated only partially and contains from 1 to 6 electrons, and these electrons are placed only on dε-AO, then we get significant energy gain.
Energy gain due to priority settlement electrons dε-atomic orbitals are called energy of stabilization of the complex by the ligand field.
The specificity of each ligand affects the field that this ligand creates - strong or weak. How stronger field ligands than more meaning splitting parameter Δ 0 .
The study of the splitting parameter is usually based on spectroscopic research. Wavelengths absorption bands complexes in a crystalline state or in solution, due to the transfer of electrons from dε- on dγ-AO, associated with splitting parameterΔ 0 as follows:
λ = c / ν; Δ 0 = E 2 – E 1 = h ν = h · ( c / λ),
where is Planck's constant h equal to 6.6260693 ∙ 10 -34 J s;
speed of light With
= 3 · 10 10 cm/s.
UnitΔ 0 is the same as the wave number: cm -1, which approximately corresponds to 12 J/mol. Splitting parameter, in addition to the type of ligand, depends on the degree of oxidation And nature complexing agent.
In complex compounds that include complexing agents of the same period and in the same oxidation state, with the same ligands, the cleavage parameter is approximately the same. With increasing degree of oxidation of the complexing agent, the value Δ 0 increases. Thus, for aqua complexes 2+ and 2+ the value of the splitting parameter is 7800 and 10400 cm -1, and for 3+ and +3 13700 and 21000 cm -1, respectively. At increasing nuclear charge of the complex-forming atom Δ 0 also increases. Hexaamminiridium(III) 3+, hexaamminiridium(III) 3+, and hexaamminiridium(III) 3+ cations ( Z= 27, 45 and 77) are characterized by splitting parameters equal to 22900, 34100 and 41000 cm -1.
The dependence of Δ 0 on the nature of the ligands is more diverse. As a result of the study of numerous complex compounds, it was found that in terms of their ability to increase the cleavage parameter of complexing metals located in their usual oxidation states, the most common ligands can be arranged in the following spectrochemical series, along which the value Δ 0 monotonically increases:
I > Br > Cl > NCS - ≈ NO 3 - > F - > OH - > H 2 O > H - > NH 3 > NO 2 - > CN - > NO > CO.
Thus, the strongest electrostatic field around the complexing agent and the strongest cleavage d-AO is caused by the ligands CN-, NO and CO. Let us consider the distribution of electrons over dε- and dγ-orbitals in the octahedral field of ligands. Check-in dε- and dγ-orbitals occurs in full accordance with Hund's rule And Pauli principle. In this case, regardless of the value of the splitting parameter, the first three electrons are occupied by quantum cells dε-sublevel:
![]() dε
dε
If the number of electrons per d- there are more than three sublevels of the complexing agent; there are two possibilities for placing them on split sublevels. At a low value of the splitting parameter (weak field of the ligands), electrons overcome the energy barrier separating dε- and dγ-orbitals; the fourth and then the fifth electrons populate quantum cells dγ-sublevel.
![]() dε
dε
With a strong ligand field and a high value of Δ 0, the population of the fourth and fifth electrons dγ-sublevel is excluded; filling in progress dε-orbitals.
![]() dε
dε
At weak field ligands populating quantum cells with 4 or 5 electrons parallel spins, so the resulting complex turns out to be strongly paramagnetic. In a strong ligand field one and then two electron pairs are formed on dε-sublevel, so paramagnetism the complex turns out to be much weaker. The sixth, seventh and eighth electrons in the case of a weak field end up back on dγ-sublevel, complementing the configurations to electron pairs (one in the case d 6, two – d 7 and three - d 8):
![]() dε
dε
In the case of a strong ligand field, the sixth electron populates dε-AO, leading to diamagnetism complex, after which the seventh and eighth electrons go to dγ-sublevel:
![]() dε
dε
Obviously, with an eight-electron configuration differences in structure between complexes with ligands weak And strong fields disappear. The occupation of orbitals by the ninth and tenth electron also does not differ for complexes of both types:
![]() dε
dε
Let us return to the consideration of the electronic structure of the octahedral complex ions 3+ and -3. According to the location in spectrochemical series, ammonia NH 3 is one of the ligands strong field, and fluoride ion F - – weak field. In the -3 anion, the F - ligands create a weak crystal field (Δ 0 = 13000 cm -1), and all electrons of the original 3 d 6 -JSC are located on dε- and dγ orbitals without any pairing. A complex ion is high-spin and contains four unpaired electrons, so it paramagnetic:

In the 3+ ion, NH 3 ligands create a strong crystal field (Δ 0 = 22900 cm -1), all 3 d 6 -electrons are placed on a more energetically favorable dε-orbitals. Transfer of electrons from dε- on dγ orbitals impossible because too high energy barrier. Therefore, this complex cation is low spin, it does not contain unpaired electrons and diamagnetic:

In a similar way, schemes for the distribution of electrons over orbitals in an octahedral field for 2+ and -4 ions can be presented:


H 2 O ligands create a weak field; exchange of electrons between dε- and dγ-orbitals does not cause any difficulties and therefore the number of unpaired electrons in the complex ion is the same as in the conventional ion Fe + II. The resulting aqua complex is high-spin, paramagnetic.
Conversely, CN - ligands cause significant cleavage d-AO, amounting to 33000 cm -1. This means that there is a strong tendency to allocate all electrons on dε-orbitals. Energy Gain, obtained with such a population of orbitals, is much greater than the energy costs due to electron pairing.
From the perspective of the valence bond method, the hybridization of valence orbitals that form a bond in the aqua complex involves d-AO external sublevel (4 sp 3 d 2), and in low-spin - d-JSC internal sublevel (3 d 2 4sp 3).
Thus, in high-spin complexes with weak-field ligands, hybridization occurs with the participation d-AO of the outer sublevel, and low-spin with high-field ligands - d- JSC of the internal sublevel. The number of unpaired electrons in the complex can be determined by electron paramagnetic resonance (EPR). Using instruments of this method, called EPR spectrometers, paramagnetic substances are studied.
The crystal field theory makes it possible to explain the appearance of one color or another in complex compounds. Among complex compounds, a significant number in the crystalline state and aqueous solution are distinguished by their bright color. Thus, an aqueous solution containing 2+ cations is colored intensely blue, 3+ cations give the solution a violet color, and 2+ cations give it a red color. If light is passed through a solution or crystalline sample of a substance visible part of the spectrum, then, in principle, three options for the physical behavior of the sample are possible: no light absorption any wavelength (substance sample colorless, although it may have absorption bands in the ultraviolet region of the spectrum); complete light absorption over the entire wavelength range (the sample will appear black); finally, light absorption only certain wavelength(then the sample will have color complementary to absorbed narrow part of the spectrum).

Thus, the color of the solution or crystals is determined frequency of absorption bands visible light. The absorption of light quanta by complexes (for example, those with an octahedral structure) is explained by the interaction of light with electrons located on dε-sublevel, accompanied by their transition to vacant orbitals dγ-sublevel. For example, when passing light through an aqueous solution containing hexaaquatitanium(III) 3+ cations, a light absorption band is detected in the yellow-green region of the spectrum (20300 cm -1, λ=500 nm). This is due to the transition of the single electron of the complexing agent from dε-AO on dγ-sublevel:

Therefore, a solution containing 3+ acquires a violet color (in addition to the absorbed yellow-green). A solution of vanadium salt Cl 3 is green. This is also due to the corresponding transitions of electrons when they absorb part of the energy of the light beam. In the ground state, with the electronic configuration of vanadium(III) 3 d 2, two unpaired electrons are occupied dε-sublevel:

There is only two options for the transition of two electrons on dγ-sublevel: either both electrons are occupied dγ-AO, or only one of them. Any other electron transitions associated with a decrease in the total spin are prohibited.
The indicated transitions of electrons that have received excess energy correspond to absorption band about 400 nm in the absorption spectrum of a solution of hexaaquavanadium(III) chloride. Absorption of the purple-violet region of the spectrum gives an additional color to the solution - bright green. If the complexing agent has an electronic configuration d 0 or d 10 then electron transitions With dε- on dγ-sublevel or vice versa impossible either because absence of electrons, either because absence of vacant orbitals. Therefore, solutions of complexes with complexing agents such as Sc(III) (3 d 0), Cu(I) (3 d 10), Zn(II) (3 d 10), Cd(II) (4 d 10), etc., do not absorb energy in the visible part of the spectrum and appear colorless. The selectivity of light absorption depends not only on complexing agent And its oxidation state, but also from type of ligands. When replacing ligands on the left side of the spectrochemical series in a complex compound with ligands that create strong electrostatic field observed increase the fraction of energy absorbed by electrons from transmitted light and, as a consequence, decrease wavelength of the corresponding absorption band. Thus, an aqueous solution containing tetraaquacopper(II) 2+ cations is blue, and a solution of tetraamminecopper(II) 2+ sulfate is intensely blue.
Related information.
