Concetto di teoria del campo cristallino. Modelli di legame chimico. Teoria del campo cristallino. Complessi a basso ed alto spin
Campo debole campo forte
Campo medio
Frac34;¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾® Δo
I ligandi a campo debole con elementi della serie 3d formano complessi ad alto spin, mentre i ligandi a campo forte formano complessi a basso spin. La differenza tra loro influisce sulla struttura elettronica dei complessi solo per le configurazioni d 4 – d 7:
3+ d5 3– d5
complesso ad alto spin complesso a basso spin
H 2 O – ligando di campo debole CN – – ligando di campo forte
I complessi a basso spin sono sempre più stabili dei complessi ad alto spin. I ligandi a campo medio, a seconda delle condizioni (carica e natura dell'atomo centrale), possono formare complessi sia ad alto che a basso spin.
Esempio. Basandosi sul TCP, formulare un'ipotesi sulla struttura elettronica degli ioni esaammina cobalto(II) (Δo = 21600 cm–1, P = 21000 cm–1) e degli ioni esaammina cobalto(III) (Δo = 9500 cm–1, P = 22500 cm–1).
L'ammoniaca è un ligando a campo medio e, a seconda del grado di ossidazione del metallo, può formare complessi sia ad alto che a basso spin. Scopriamo quali complessi saranno energeticamente più stabili per il cobalto(II) e il cobalto(III). Per fare ciò, confronta l'ESC di ciascuno ione in un campo forte e debole:
(a) 3+, d 6

campo forte campo debole
ESKP (campo forte) = –6´(2/5)Δo + 2P = –6´(2/5) ´21600 + 2´21000 = –9840 cm –1
ESKP (campo debole) = –4´(2/5)Δo + 2´(3/5)Δo = –4´(2/5) ´21600 + 2´(3/5) ´21600 = –8640 cm – 1
Il guadagno energetico è maggiore nel caso di un complesso a basso spin.
(b) 2+, d7

campo forte campo debole
ESKP (campo forte) = –6´(2/5)Δo + 1´(3/5)Δo + P = –6´(2/5)´9500 + 1´(3/5) ´9500 + 22500 = 7900 cm–1
ESKP (campo debole) = –5´(2/5)Δo + 2´(3/5)Δo = –5´(2/5) ´9500 + 2´(3/5) ´9500 = –7600 cm – 1
Il guadagno energetico è maggiore nel caso di un complesso ad alto spin.
Pertanto, lo ione 3+ è a basso spin e lo ione 2+ è ad alto spin.
L'ESC aumenta con l'aumentare di Δo, tuttavia, è diverso per gli stati ad alto e basso spin (Fig. 1.28. La dipendenza dell'ESC per complessi ad alto e basso spin con configurazione d 6 dal valore Δo = 10Dq La regione in cui è possibile l'esistenza di entrambi gli Stati è ombreggiata). La regione vicino al punto di intersezione di queste due linee corrisponde a complessi che possono esistere sia negli stati ad alto spin che in quelli a basso spin.
Un esempio è il complesso tiocianato di ferro (II) con 1,10-fenantrolina, che è ad alto spin (paramagnetico) a basse temperature e a basso spin (diamagnetico) a temperature elevate (M. Marchivie, P. Guionneau, J. A. K. Howard , G. Chastanet, J.-F. Letard, A. E. Goeta, D. Chasseau, J. Am. Chem. Soc., 2002, v. 124, pagina 194). Il cambiamento nella molteplicità è accompagnato da un cambiamento nelle distanze interatomiche e nella geometria dell'ambiente di coordinazione: il complesso a basso spin è un ottaedro regolare, mentre il complesso ad alto spin è distorto. La transizione inversa allo stato di spin elevato è possibile sotto l'influenza di alte pressioni o radiazioni. Attualmente sono note diverse dozzine di sistemi di questo tipo.
Parlando delle proprietà σ-donatore e π-accettore del ligando, siamo andati oltre il TCP, utilizzando gli approcci del metodo orbitale molecolare applicato a composti complessi (Volume 1). Ricordiamo che l'immagine della scissione degli orbitali d è un frammento dello schema generale degli orbitali molecolari in un complesso ottaedrico, dove gli orbitali t 2g sono considerati non leganti ed e g - come antilegame (Fig. Volume 1) .
La formazione di legami in un complesso ottaedrico senza legame π coinvolge gli orbitali s, p e d del metallo e un orbitale di ciascun ligando. Da 15 orbitali atomici si formano 15 orbitali molecolari, sei dei quali (a 1 g, t 1 u, e g (nota a piè di pagina: la lettera nella designazione degli orbitali indica il grado della loro degenerazione: t - tre volte degenere, e - doppiamente degenerato, a - non degenerato e la presenza di un centro di simmetria: g - simmetrico, u - asimmetrico)) σ-legame, tre (t 2 g) - non legame e sei (e g *, t 1 u *, un allentamento σ di 1 g *). Gli orbitali di legame sono più vicini in termini di energia agli orbitali del ligando, mentre gli orbitali di non legame sono localizzati prevalentemente sull'atomo di metallo. L'energia d xy , d xz , d yz (t 2 g) degli orbitali metallici praticamente non cambia durante la formazione del complesso.
La presenza di un orbitale vacante a bassa energia nel ligando, simile per simmetria agli orbitali metallici, porta ad una diminuzione dell'energia degli orbitali t 2g, praticamente senza influenzare eg, aumentando così Δо (Fig. 1.29. Frammenti del MO diagramma per il complesso di cobalto (III) con ligando donatore σ (a) e ligando donatore σ, ligando π (b)).
Effetto Jahn-Teller. Nel 1937 Yang e Teller dimostrarono il teorema secondo il quale qualsiasi molecola non lineare in uno stato elettronico degenere è instabile e subisce spontaneamente una distorsione che ne abbassa la simmetria e porta alla rimozione della degenerazione. Il teorema prevede solo il fatto stesso di rimuovere la degenerazione, ma non indica come verrà rimossa. Sulla base di questo teorema, fu spiegata la distorsione della geometria ottaedrica di un numero di complessi e il fatto stesso della presenza di tale distorsione fu chiamato effetto Jahn-Teller. Diamo un'occhiata a un esempio. I complessi di rame(II) con la configurazione d9, di regola, non rappresentano un ottaedro regolare, ma sono allungati o compressi lungo uno degli assi (Fig. 1.30. Distorsione della geometria ottaedrica nei complessi di rame(II)). Consideriamo il caso di un ottaedro prolato. La rimozione dei ligandi situati lungo l'asse z provoca la rimozione della degenerazione dovuta a un cambiamento nelle energie degli orbitali. Gli orbitali diretti lungo l'asse z (d xz, d yz, d z 2) interagiscono più debole con gli orbitali dei ligandi rispetto agli orbitali che non hanno una componente z (d xy, d x 2 -y 2), e quindi abbassano la loro energia. Una coppia di orbitali della stessa simmetria, aventi una componente z (d xz, d yz), rimane degenere e acquisisce maggiore energia. (Fig. 1.31. Cambiamento nelle energie degli orbitali d quando l'ottaedro è distorto). L'effetto Jahn-Teller si manifesta più fortemente nei complessi con orbitali e g riempiti in modo diseguale, cioè con configurazioni t 2g 3 e g 1 (corrispondente allo ione d 4 in un campo debole: CrCl 2, K 3 MnF 6) e t 2g 6 e g 3 (corrisponde allo ione d 9: quasi tutti i complessi di rame(II)) e t 2g 6 e g 1 (corrisponde allo ione d 7 in un campo forte, raro, K 3 NiF 6). Un effetto Jahn-Teller insignificante è tipico per i complessi con orbitali t 2g riempiti in modo diseguale, cioè per configurazioni elettroniche t 2g 1 (d 1), t 2g 2 (d 2), t 2g 4 (d 4 in un campo forte) , t 2g 5 (d 5 in campo forte), t 2g 5 e g 1 (d 6 in campo debole), t 2g 5 e g 2 (d 7 in campo debole). Gli ioni con le configurazioni d 3 e d 5 in campo debole, d 3 e d 6 in campo forte, d 8 e d 10 non sono in nessun caso Jahn-Teller.
L'effetto Jahn-Teller si manifesta nella disuguaglianza delle lunghezze dei legami in molti complessi di rame (II) e manganese (III) e in un cambiamento non monotono nelle costanti di stabilità graduale dei complessi. Ad esempio, nel cloruro di rame anidro (II), l'atomo di rame è circondato da sei atomi di cloro, quattro dei quali si trovano a una distanza di 0,230 nm e gli altri due a una distanza di 0,295 nm da esso.
Sono noti complessi di rame(II) (Cl 2, (C 6 H 5 SO 3) 2, ecc.), costituiti da più ioni Jahn-Teller cristallograficamente non equivalenti, ciascuno con il proprio tipo di distorsione, che si trasformano l'uno nell'altro, cambiando la distanza metallo-legante è così veloce che nel complesso tutte le distanze metallo-legante sembrano essere le stesse. Questo caso è stato chiamato effetto Jahn-Teller dinamico o pulsante(P.E.M. Wijnands, J.S. Wood, J. Redijk, W.J.A. Maaskant, Inorg. Chem., 1986, 35, 1214).
L’effetto Jahn-Teller, tuttavia, non è una legge universale. Attualmente sono noti ioni complessi con configurazione Jahn-Teller, che sono ottaedri non distorti: 4–, 3+.
Divisione in campi con simmetria diversa da quella ottaedrica.
Oltre a quelli ottaedrici, sono noti molti complessi con una geometria diversa: piano quadrato, tetraedrico, piramidale trigono, piramidale quadrato, lineare, ecc. La suddivisione in ciascuno di questi campi è diversa rispetto all'ottaedro; è determinato dalla simmetria del poliedro di coordinazione.
I complessi quadrato-planari possono essere considerati un caso estremo di distorsione tetragonale della geometria ottaedrica, quando i ligandi situati lungo uno degli assi delle coordinate vengono rimossi all'infinito (Fig. 1.27b). Le designazioni degli orbitali sono mostrate in figura. I complessi planari-quadrati sono più tipici per gli ioni con la configurazione elettronica d 8 – Ni 2+, Pd 2+, Pt 2+, Au 3+. La loro stabilità aumenta notevolmente all'aumentare di Δ, cioè quando si passa da elementi della serie 3d a elementi di transizione pesanti. Quindi, ad esempio, se palladio, platino e oro hanno quasi tutti complessi con un numero di coordinazione pari a quattro quadrati, allora il nichel forma complessi planari-quadrati solo con ligandi ad alto campo: 2–, Ni(dmg) 2. I complessi di nichel (II) con ligandi a basso campo, come gli alogeni, hanno una geometria tetraedrica.
Alcuni complessi di metalli di transizione planari quadrati formano catene in forma solida con ligandi a ponte, ad esempio Pt-CN-Pt in K 2 Br 0,3, dove gli atomi di platino sono parzialmente nello stato di ossidazione +4. L'elevata capacità di penetrazione degli orbitali 5d garantisce la loro sovrapposizione con la formazione di un'unica banda energetica e, di conseguenza, conduttività metallica nella direzione della catena. Tali complessi molecolari sono in grado di condurre corrente elettrica e sono attualmente oggetto di studi approfonditi.
In un campo di simmetria tetraedrica, gli orbitali d xy , d yz , d xz hanno l'energia massima, sono chiamati orbitali t 2, e l'energia minima è gli orbitali d x 2 –y 2 e d z 2, sono indicati e . A causa della presenza di un minor numero di leganti e della loro diversa disposizione, il campo tetraedrico (Fig. 1.32. Confronto delle scissioni nei campi tetraedrico e ottaedrico) risulta essere 2,25 volte più debole di quello ottaedrico: .
La maggior parte dei complessi tetraedrici sono ad alto spin. (Nota a piè di pagina - Sono noti diversi esempi di complessi tetraedrici a basso spin, ad esempio Cr(N(Si(CH 3) 3) 2 ) 3 NO (cromo(II), d 4 ; D. C. Bradley, Chem. Ber., 1979, 11, 393); CoL 4, dove L è 1-norbornile (cobalto(IV), d 5; E. K: Brune, D. S. Richeson, K. H. Theopold, Chem. Commun., 1986 , 1491)). La massima stabilizzazione dell'ambiente tetraedrico da parte del campo cristallino si ottiene con le configurazioni d 2 (FeO 4 2–, MnO 4 3–) e d 7 (2–). A causa dell'energia di stabilizzazione relativamente bassa, i complessi tetraedrici sono più spesso formati da ioni con configurazioni d 0 (TiCl 4, MnO 4 –, CrO 4 2–), d 5 in un campo debole (FeCl 4 –) e d 10 (ZnCl 4 2–) con zero ESKP, nonché ioni di metalli non di transizione (AlCl 4 –). La formazione di complessi tetraedrici rispetto a quelli ottaedrici è spesso favorita dal fattore sterico, ad esempio lo ione è più stabile del 3–.
Usare TCP per spiegare la stabilità dei complessi. Serie Irving-Williams. La teoria del campo cristallino consente di spiegare la natura non monotona dei cambiamenti nelle energie del reticolo cristallino di ossidi e alogenuri, costanti di stabilità dei complessi, ecc. L'ordine di cambiamento nelle energie di idratazione dei cationi a doppia carica di metalli 3d generalmente coincide con la natura dei cambiamenti nell'ESC nei complessi ad alto spin (Fig. 1.33. Cambiamento nell'energia di idratazione dei metalli cationici a doppia carica della serie 3d (a) e cambiamento nell'ESC nei complessi ad alto spin (b) ), quanto più forte è la stabilizzazione da parte del campo cristallino, tanto maggiore è l'idratazione. È noto che le costanti di sostituzione di una molecola d'acqua con un ligando a campo debole L
2+ + L x– = (2-x)+ + H 2 O
obbedire alla serie Irving-Williams: Mn 2+< Fe 2+ < Co 2+ < Ni 2+ < Cu 2+ < Zn 2+ (Рис. 1.34. Зависимость первой константы устойчивости комплекса от природы 3d-металла). Согласно этому ряду, наибольшей устойчивостью обладают комплексы меди(II) и никеля(II). Простейший вариант ЭСКП предсказывает наибольшую устойчивость никелевых комплексов. При этом надо учитывать, что комплексы меди(II) имеют сильно искаженную октаэдрическую геометрию, что вносит существенный вклад в величину константы устойчивости.
Effetto nefeloauxetico. Si è scoperto che la mutua repulsione degli elettroni d si indebolisce quando l'atomo si trova nel campo dei ligandi. Questo effetto del ligando sugli elettroni d dell'atomo di metallo è chiamato effetto nefeloauxetico dalle parole greche νεφελη - nuvola e αυξανω - aumento. La serie dei ligandi, disposti in ordine crescente di influenza sugli orbitali metallici, corrisponde quasi completamente alla serie spettrochimica. La ragione dell'effetto nefeloaxetico è la sovrapposizione degli orbitali D del metallo con gli orbitali dei ligandi, grazie alla quale la nuvola D si espande nello spazio. La presenza di questo effetto dimostra chiaramente i limiti del modello elettrostatico più semplice: la teoria del campo cristallino, che presuppone che i lignati siano cariche negative puntiformi.
Teoria dei campi dei leganti. La teoria del campo cristallino è stata sviluppata da Bethe nel 1929. Attualmente è ampiamente utilizzata in una forma modificata con correzioni per una certa covalenza del legame metallo-ligando. Questa teoria è chiamata teoria del campo del ligando. La presenza di un contributo covalente modifica l'energia degli orbitali metallici rispetto a quella calcolata dal TCP. La proporzione di covalenza viene presa in considerazione introducendo fattori di correzione che consentono di equiparare i valori sperimentali a quelli calcolati.
Colorazione dei complessi.
Il colore dei complessi degli elementi di transizione d è associato alle transizioni elettroniche da un orbitale d all'altro. Ciò è chiaramente illustrato dall'esempio dello ione Ti 3+, discusso nel primo volume del libro di testo. Assorbendo energia corrispondente alle parti blu e verde dello spettro visibile, l'unico elettrone d nello ione Ti 3+ si sposta nell'orbitale e g (Fig. 1.35. Spettro dello ione 3+). Il colore dello ione è dovuto a colori aggiuntivi: rosso e viola. (Nota a piè di pagina - Il lettore attento noterà una certa asimmetria della banda di assorbimento. È una conseguenza di una leggera suddivisione del livello t 2g causata dall'effetto Jahn-Teller). Nel secondo risguardo del libro di testo è presentato un diagramma che mostra i colori complementari e che è ben noto a ogni artista. L'energia di transizione, espressa in centimetri reciproci (1000 cm –1 = 12 kJ), corrisponde al parametro di scissione Δο - nella maggior parte dei casi è determinata dagli spettri elettronici. La lunghezza d'onda è inversamente proporzionale all'energia:
 .
.
Nel caso di complessi con un gran numero di elettroni, l'immagine dello spettro diventa più complicata e in esso compaiono bande aggiuntive. Ciò è dovuto al fatto che lo stato eccitato t 2g 1 e g 1 può essere realizzato in diversi modi, a seconda di quali due orbitali d si trovano gli elettroni. Ad esempio, uno stato in cui gli elettroni occupano gli orbitali d xy e d x 2 –y 2 avrà un'energia maggiore rispetto a uno stato d xy 1 d z 2 1 a causa della maggiore repulsione degli elettroni lungo l'asse x. L'energia corrispondente alla banda con l'energia più bassa è pari al parametro di splittaggio Δo.
Per descrivere più nel dettaglio gli spettri elettronici è necessario introdurre alcuni concetti. Chiamiamo microstato qualsiasi disposizione di elettroni a un sottolivello. Il numero di microstati N, in cui n elettroni occupano x orbitali, è uguale a

Ogni microstato è caratterizzato da propri valori di spin e momento angolare. Viene chiamato un insieme di microstati con energie identiche termine, ad esempio, 3 P, 5 D, 1 S. L'indice digitale indica la molteplicità, che viene calcolata come:
molteplicità = numero di elettroni spaiati nello stato fondamentale + 1.
I nomi dei termini si leggono con indicazione di molteplicità: “tripletta P”, “quintetto D”, “singolo S”. La lettera indica il momento angolare totale L di un atomo o ione, che è uguale al valore massimo della somma dei momenti angolari m l dei singoli orbitali occupati dagli elettroni. Ad esempio, lo ione Ti 3+ contiene un elettrone d, il numero di microstati è N = (2´5)!/1!(2´5 – 1)! = 10, L = 2(D) (poiché per l'orbitale d m l = –2, –1, 0, 1, 2 il numero di elettroni è 1, quindi la somma massima m l è pari al valore più grande di m l), molteplicità 1 + 1 = 2. Pertanto, il termine dello stato fondamentale (con l'energia più bassa) è 2 D. Nel caso di uno ione con configurazione elettronica d 2 N = (2´5)!/2!( 2´5 – 2)! = 45, L = 3(F) (poiché per l'orbitale d m l = –2, –1, 0, 1, 2 il numero di elettroni è 2, quindi la somma massima dei due valori maggiori è uguale a m l), molteplicità 2 + 1 = 3. Di conseguenza, il termine del microstato fondamentale è 3 F. Con una diversa disposizione di due elettroni sul sottolivello d si ottengono stati descritti con altri termini: 3 P, 1 G , 1 D, 1 S, ecc. La relazione tra i valori numerici di L e i simboli alfabetici è riportata di seguito:
L = 0 1 2 3 4 5 6 7
Allo stesso modo, possiamo derivare i termini degli stati fondamentale ed eccitato per altri ioni di elementi d (Tabella 1.5.). Si noti che i termini per gli ioni con configurazione d n e d 10-n sono gli stessi.
Tavolo. 1.5.
Termini degli stati fondamentali e stati eccitati più vicini per varie configurazioni di elettroni d.
I termini sono divisi nel campo ottaedrico come orbitali, indicati con lettere simili. I termini D sono suddivisi in componenti T 2 g ed E g, come gli orbitali d, i termini F - in T 1 g, T 2 g e A 2 g, come gli orbitali f. I termini S e P non sono affatto divisi. Le possibilità di transizioni elettroniche tra stati diversi sono limitate dalle regole di selezione. Pertanto, nei complessi sono consentite solo transizioni tra stati con la stessa molteplicità. Ciascuna di queste transizioni corrisponde ad una banda nello spettro di assorbimento. Ad esempio, consideriamo lo spettro elettronico del complesso 3+ (Fig. 1.36. Spettro elettronico del complesso 3+). Le tre bande sono dovute a tre transizioni elettroniche: 4 A 2 g ® 4 T 2 g, 4 A 2 g ® 4 T 1 g, 4 A 2 g ® 4 T 1 g (P). La transizione con l'energia più bassa corrisponde al valore del parametro di splittaggio: Δo = 17400 cm–1. Il complesso assorbe la luce nelle parti rossa (17400 cm–1) e blu (23000 cm–1) dello spettro visibile e nel vicino ultravioletto (37800 cm–1), pertanto ha un colore viola.
Secondo la regola di Laporte, le transizioni tra stati con la stessa parità, che includono transizioni s-s, p-p, d-d, f-f, sono improbabili o, nel linguaggio della spettroscopia, sono vietate nei complessi ottaedrici. Le transizioni proibite sono possibili, ma si verificano con bassa intensità. Questo è il motivo per cui i sali dei metalli di transizione hanno un colore evidente solo in soluzioni concentrate. È molte volte più debole del colore del permanganato o del dicromato, i cui ioni non contengono elettroni d.
La regola di Laporte è applicabile solo nel caso di complessi che hanno un centro di simmetria. Quando l’ottaedro viene distorto, il centro di simmetria scompare, il divieto di Laporte viene revocato e appare il colore. Ad esempio, lo ione 3+ è incolore, ma le soluzioni di sali di ferro (III) sono spesso giallo-arancio a causa dell'idrolisi che porta alla formazione di particelle asimmetriche con un ambiente ottaedrico distorto.
Il colore dei complessi, oltre alle transizioni d-d da un orbitale d metallico a un altro (da t 2g a e g nei complessi ottaedrici), è determinato da altri due fattori: transizioni dagli orbitali del ligando agli orbitali metallici (sono chiamati trasferimento di carica ) e transizioni all'interno degli orbitali del ligando. Queste transizioni non rientrano nella regola di Laporte e, quindi, hanno un'elevata intensità.
La banda di trasferimento di carica è presente nello spettro elettronico di qualsiasi composto, tuttavia in alcuni casi si trova nella parte ultravioletta dello spettro e non viene da noi percepita come colore. Se la differenza tra le energie degli orbitali del ligando e degli orbitali del metallo si riduce, la banda di trasferimento di carica cade nella parte visibile dello spettro. È il trasferimento di carica che spiega il colore intenso del perossido di permanganato, del dicromato, del solfuro di mercurio, del titanio (IV) e di molti altri composti con orbitali d vuoti. In alcuni casi, sotto l'influenza della luce, il trasferimento di carica dagli orbitali del ligando agli orbitali del metallo avviene in modo irreversibile, cioè è accompagnato da un processo chimico. Un esempio è la decomposizione fotochimica degli alogenuri d'argento, che è alla base della fotografia in bianco e nero: Ag + Br – ¾® Ag 0 + Br 0 .
Nello spettro elettronico del permanganato di potassio si osservano quattro bande, corrispondenti alle transizioni di elettroni da orbitali di non legame localizzati prevalentemente sul ligando (orbitali a 1, t 2 σ e orbitali e, t 1, t 2 π) a e*, t2 '' orbitali antilegame orbitali localizzati sull'atomo di metallo ((Fig. 1.37. Diagramma energetico dello ione tetraedrico MnO 4 - con legame π. Le transizioni elettroniche sono mostrate dalle frecce):
ν 1 , Mn(e*) ¾ O(t 1) 17700 cm –1
ν 2 , Mn(t 2 '') ¾ O(t 1) 29500 cm –1
ν 3 , Mn(e*) ¾ O(t 2) 30300 cm –1
ν 4 , Mn(t 2 '') ¾ O(t 2) 44400 cm –1
La banda con l'energia più bassa cade nella parte visibile dello spettro (λ = 107/17700 = 565 nm), che corrisponde all'assorbimento della luce verde e alla trasmissione della luce rosso-cremisi.
3. Meccanismi di reazioni che coinvolgono composti complessi.
La stragrande maggioranza dei processi chimici avviene come una catena sequenziale di alcuni stadi elementari e l'equazione di reazione contiene solo informazioni sui principali prodotti finali della reazione. Questa sequenza di trasformazioni elementari nel percorso dalle sostanze di partenza ai prodotti è chiamata meccanismo. I composti intermedi, solitamente instabili, attraverso i quali scorre il percorso dai reagenti ai prodotti, sono chiamati intermedi. Qualsiasi intermedio ha una certa durata, solitamente estremamente breve, fino a 10 -14 s. Sul profilo energetico della reazione corrisponde ad un minimo (Fig. a) (Fig. 1.38. Profili energetici di una reazione che procede attraverso: (a) intermedio, (b) stato di transizione.). Di norma gli intermedi possono essere rilevati in una miscela di reazione mediante metodi spettrali e solo in rari casi possono essere isolati in forma individuale. Pertanto, le informazioni principali sul meccanismo di reazione si ottengono solitamente studiandone la cinetica, determinando le costanti di velocità e calcolando i parametri di attivazione (entalpia, entropia, volume). In questo caso il meccanismo è un modello conforme ai dati cinetici, un modello che può essere migliorato, modificato, rivisto.
In alcune reazioni, gli intermedi non si formano e la transizione dai reagenti ai prodotti avviene in sequenza: uno degli atomi viene gradualmente rimosso e l'altro si avvicina. In questo caso si dice che la reazione procede stato di transizione o complesso attivato. Corrisponde ad un massimo nel profilo energetico della reazione (Fig. B).
Aggiunta: Complessi labili e inerti
La stabilità termodinamica di una particella è determinata dalla variazione dell'energia di Gibbs per la reazione della sua dissociazione, o dal valore della costante di stabilità di questo processo. La stabilità cinetica mostra la velocità con cui una data particella interagisce con altre particelle o subisce un decadimento. Viene considerata una particella chimica inerte, se reagisce con un'emivita superiore a 1 minuto. Vengono chiamate le particelle che reagiscono ad una velocità più elevata labile. Va ricordato che la stabilità cinetica e termodinamica non dipendono l'una dall'altra, cioè la stessa sostanza può avere un'elevata costante di stabilità e allo stesso tempo essere inerte o, al contrario, labile. Alcuni di questi esempi sono riportati nella Tabella 1.6.
Tabella 1.6. Costanti di stabilità e velocità di sostituzione dei ligandi nei cianocomplessi di alcuni metalli.
Henry Taube ha mostrato la connessione tra la stabilità cinetica dei complessi ottaedrici e la configurazione elettronica dello ione centrale nel campo ottaedrico. Secondo Taube i seguenti complessi sono labili:
· possedere almeno un orbitale t 2g vacante - possono usarlo nelle reazioni secondo il meccanismo associativo (A, I a), oppure
· avere almeno un elettrone nell'orbitale e g - questo favorisce la reazione con il meccanismo dissociativo (D, I d), perché La rimozione di un elettrone dall'orbitale eg abbassa l'energia dello stato di transizione.
Pertanto, i complessi ottaedrici di cromo(III) (t 2g 3), i complessi a basso spin di ferro(II) (t 2g 6) e ferro(III) (t 2g 5), nonché i complessi di 4d-, 5d- gli elementi di transizione sono classificati come inerti con il numero di elettroni d superiore a due.
FINE DELL'APPENDICE
Una classificazione unificata delle reazioni inorganiche non è stata ancora sviluppata. Convenzionalmente si può proporre il seguente schema (Fig. 1.39. Schema illustrante la classificazione delle reazioni inorganiche):
1) Le reazioni di sostituzione, aggiunta o eliminazione dei ligandi influenzano un cambiamento nella sfera di coordinazione del metallo,
2) Le reazioni redox sono associate a un cambiamento nella configurazione elettronica del metallo, ma non influenzano il suo ambiente di coordinazione,
3) Le reazioni dei ligandi coordinati comportano un cambiamento nel ligando senza cambiare la sfera di coordinazione del complesso.
Reazioni di sostituzione. In senso lato, per reazioni di sostituzione si intendono i processi di sostituzione di alcuni ligandi nella sfera di coordinazione di un metallo con altri. Tali reazioni possono avvenire con o senza cambiamento dello stato di ossidazione. Seguendo la classificazione di cui sopra, useremo questo termine solo in relazione a reazioni che avvengono senza cambiamento degli stati di ossidazione.
La classificazione delle reazioni di sostituzione in chimica inorganica è stata sviluppata da Langford e Gray. Si basa sulla definizione del cosiddetto meccanismo limitante e non sulla descrizione di un meccanismo specifico. Innanzitutto viene determinato il meccanismo stechiometrico e poi quello interno. Meccanismo stechiometricoè una sequenza di fasi elementari nel passaggio dalle sostanze di partenza ai prodotti. Può essere dissociativo (D), associativo (A) e di scambio (scambio reciproco, I). I processi dissociativi e associativi rappresentano, per così dire, due casi limite, direttamente opposti l'uno all'altro. Entrambi i processi avvengono in due fasi attraverso la formazione di un intermedio.
Dissociativo (D)
Il processo è a due stadi, nel caso limite procede attraverso un intermedio a concentrazione ridotta:
ML 6 + L, + Y ¾® ML 5 Y
Associativo (A)
Il processo è a due fasi, caratterizzato dalla formazione di un intermedio con una maggiore concentrazione:
ML 6 + Y, ¾® ML 5 Y + L
Scambio reciproco (I)
La maggior parte delle reazioni di scambio procedono attraverso questo meccanismo. Il processo è in una fase e non è accompagnato dalla formazione di un intermedio. Nello stato di transizione, il reagente e il gruppo uscente sono associati al centro di reazione, entrano nella sua sfera di coordinazione più vicina e durante la reazione un gruppo viene spostato da un altro, avviene uno scambio di due ligandi:
ML 6 + Y ML 5 Y + L.
Lo stato di transizione è un complesso della sfera esterna o, nel caso di ligandi carichi, una coppia ionica MX 5 L + Y - .
Meccanismo interno (UN O D) caratterizza il processo di sostituzione del ligando a livello molecolare. Mostra quale dei due processi – la formazione o la rottura di un legame nello stato di transizione – è limitante. Se la velocità di reazione è determinata dalla formazione di un legame tra il centro di reazione e il reagente, si parla di attivazione associativa. Altrimenti, quando il fattore limitante è la rottura della connessione tra il centro di reazione e il gruppo uscente, il processo procede con attivazione dissociativa. Passando al meccanismo stechiometrico, è facile notare che il processo dissociativo corrisponde sempre all'attivazione dissociativa, e il processo associativo corrisponde sempre all'attivazione associativa, cioè il concetto di meccanismo interno risulta informativo solo nel caso di un meccanismo di scambio reciproco: può verificarsi sia con attivazione dissociativa (I d) che associativa (I a). Nel caso del meccanismo di scambio reciproco con attivazione associativa (Ia), la velocità di reazione dipende dalla natura di Y. Nello stato di transizione, l'atomo di metallo è strettamente legato sia al gruppo uscente che al nucleofilo attaccante. Un esempio è il processo di sostituzione di un atomo di cloro con bromo e iodio in un complesso di platino con dietilenetriamina (dien):
Y - ¾¾® + + Cl -
Y = Br, le velocità I variano notevolmente.
Nel caso del meccanismo di scambio reciproco con attivazione dissociativa (I d), la velocità di reazione non dipende dalla natura del reagente Y. I gruppi attaccanti e uscenti nello stato di transizione sono debolmente legati allo ione centrale. Questo meccanismo viene utilizzato per sostituire l'acqua con l'ammina negli acquacomplessi di molti metalli di transizione, ad esempio il nichel:
2+ + Y¾¾® 2+ + H2O
Y = NH 3 , le velocità py sono vicine.
Lo studio dei meccanismi delle reazioni di sostituzione nei complessi di molti metalli è solo nella fase iniziale. Informazioni complete sono state ottenute solo per i complessi quadrato-planari di platino e per i complessi ottaedrici di cromo (III) e cobalto (III). Si può considerare fermamente stabilito che nei complessi di platino (II) la sostituzione avviene secondo il meccanismo associativo (A, Ia) attraverso uno stato intermedio o di transizione sotto forma di una bipiramide trigonale. I complessi ottaedrici di cobalto (III) reagiscono in modo dissociativo (meccanismi D, I d). Esempi specifici di tali reazioni verranno presi in considerazione quando si descriverà la chimica di questi elementi.
Reazioni redox. La maggior parte dei processi redox sono una combinazione complessa di singoli stadi elementari, ciascuno dei quali comporta il trasferimento di uno o, molto meno frequentemente, di due elettroni. Il trasferimento simultaneo di un numero maggiore di elettroni nelle soluzioni è impossibile.
Il trasferimento di un singolo elettrone può avvenire attraverso uno dei due meccanismi: sfera esterna, cioè mediante tunneling, o sfera interna, attraverso un ligando a ponte. Il meccanismo intrasfera è realizzato in complessi contenenti alogenuri, ioni idrossido e gruppi carbossilici che possono fungere da ponti tra i metalli. Un esempio è la reazione tra gli ioni pentammina clorocobalto (III) ed esaacquacromo (II). Il processo può essere approssimativamente suddiviso in tre fasi: la formazione di un complesso eterometallico con uno ione cloruro a ponte, il trasferimento di elettroni e la decomposizione del complesso a ponte. Lo ione 2+ risultante, essendo labile, si trasforma istantaneamente in un acquacomplesso e l'inerte [(H 2 O) 5 CrCl] 2+ non interagisce con l'acqua:

Se nel sistema non ci sono particelle che possano fungere da ponti, il processo procede nella sfera esterna:
2+ + 3+ = 3+ + 2+ .
È particolarmente necessario evidenziare le reazioni di addizione ossidativa ed eliminazione riduttiva, discusse nel capitolo 6.
Reazioni di ligandi coordinati. Questo gruppo di reazioni comprende processi di modifica dei ligandi coordinati da uno ione metallico. Ad esempio, i complessi dichetonici, come i dichetoni liberi, possono essere nitrati, acilati o alogenati. L'esempio più interessante e insolito di reazioni di ligandi coordinati è sintesi del modello– un metodo unico per “assemblare” un ligando su uno ione metallico. Un esempio è la sintesi delle ftalocianine dal nitrile dell'acido ftalico, che avviene in presenza di ioni rame (II), e la sintesi di una base di Schiff macrociclica dalla 2-amminobenzaldeide, che avviene in presenza di ioni nichel (II):

In assenza di metallo il processo procede lungo un percorso diverso e il prodotto desiderato è presente solo in piccola quantità nella miscela di reazione. Lo ione metallico agisce nella sintesi del modello come una matrice (“modello”), stabilizzando uno dei prodotti che sono in equilibrio tra loro e spostando l'equilibrio verso la sua formazione. Ad esempio, nella reazione X + Y ¾® si forma una miscela di prodotti A e B, in cui prevale B, che ha un'energia inferiore. In presenza di uno ione metallico, la sostanza A predomina nei prodotti di reazione sotto forma di un complesso con M (Fig. 1.40. Diagramma energetico dell'interazione di X e Y in assenza di uno ione metallico (a sinistra) e nella sua presenza (b)).
Domande e compiti
1. Quale dei seguenti composti ha una struttura perovskite? BaTiO 3, LiNbO 3, LaCrO 3, FeTiO 3, Na 2 WO 4, CuLa 2 O 4, La 2 MgRuO 6. La tabella dei raggi ionici è riportata in Appendice. Tieni presente che nelle fasi di ossido complesso, le posizioni B possono contenere cationi di due metalli diversi.
2. Utilizzando il TCP, determinare se i seguenti spinelli saranno diritti o invertiti: ZnFe 2 O 4, CoFe 2 O 4, Co 3 O 4, Mn 3 O 4, CuRh 2 O 4.
3. Ione tiocianato SCN - ha due centri donatori: duro e morbido. Prevedere quale struttura avranno i complessi tiocianici di calcio e rame(I). Perché non è possibile ottenere tiocianato di rame (II)?
4. Lo spettro dell'acquaione Cr 2+ (termine dello stato fondamentale 5 D) ha due bande (Fig. 1.41. Spettro dell'acquaione Cr 2+), sebbene tra i termini degli stati eccitati più vicini non ce n'è uno con la stessa molteplicità. Cosa spiega questo? Di che colore ha questo ione?
5. Utilizzando i valori Δο di seguito, calcolare l'ESC per i seguenti complessi in kJ/mol:
(a) 2–, Δο = 15000 cm–1,
(b) 2+, Δο = 13.000 cm–1,
(c) 2–, Δο (per 4–)= 21000 cm–1,
Prendi l'energia di accoppiamento pari a 19000 cm –1, 1 kJ/mol = 83 cm –1. Calcolare i loro momenti magnetici (componente di spin).
6. Utilizzando TCP, spiegare perché lo ione CN – reagisce con lo ione esaquanickel(III) per formare esacianoferrato(II) e con lo ione esaquanickel(II) per formare tetracianonickelato(II).
7. Di seguito sono riportate le costanti di reazione per la sostituzione sequenziale dell'acqua nell'acquacomplesso di rame(II) con ammoniaca: K 1 = 2´10 4 , K 2 = 4´10 3 , K 3 = 1´10 3 , K 4 = 2´10 2 , K5 = 3´10 –1, K6<< 1. Чем объясняется трудность вхождения пятой и шестой молекул аммиака в координационную сферу меди?
8. Come cambia la rigidità dei cationi quando si muovono lungo una fila 3d? Ciò è coerente con l'ordine di cambiamento delle costanti di stabilità dei complessi (serie di Irving-Williams, Fig. 1.34).
9. Spiega perché lo ione ferro esaquatico (III) è incolore e le soluzioni di sali di ferro (III) sono colorate.
10. Suggerire un meccanismo per la reazione 3– + 3– = 4– + 2–, se è noto che l'introduzione dello ione tiocianato nella soluzione porta ad un cambiamento nella velocità di reazione, e la velocità è praticamente indipendente dalla presenza di ammoniaca. Offri una spiegazione per questi fatti.
Il concetto di cambiamento nella struttura elettronica degli ioni dei metalli di transizione sotto l'azione del campo elettrico delle particelle cariche che li circondano è stato proposto da Becquerel e ulteriormente sviluppato da H.A. Bethe e J. Van Vleck all'inizio XX V. Questi concetti sono stati applicati alla descrizione della struttura elettronica e delle proprietà dei composti complessi solo a metà XX secolo da H. Hartmann e il modello fu chiamato “teoria del campo cristallino” (CFT).
Disposizioni fondamentali del TCH per i complessi di transizione d metalli Fig. 24):
1. - Il complesso esiste ed è stabile grazie all'interazione elettrostatica dell'agente complessante con i ligandi.
2. - I ligandi vengono considerati senza tener conto della loro struttura elettronica come cariche puntiformi o dipoli.
3. - Sotto l'influenza del campo elettrico dei ligandi, la valenza quintuplica degenera ( n-1) d gli orbitali sono divisi a seconda della simmetria dell'ambiente del ligando.
4. - Distribuzione degli elettroni di valenza dei metalli tra gli split ( n-1) d orbitali dipende dal rapporto tra l'energia di accoppiamento degli spin e l'energia di scissione.
Consideriamo, ad esempio, il cambiamento nell'energia di cinque volte degenere ( n-1) d orbitali dello ione metallico centrale M n+ , situato al centro delle coordinate, sotto l'influenza del campo ottaedrico di ligandi caricati negativamente [ ML 6] z , situato sugli assi delle coordinate (Fig. 25). Come risultato della repulsione degli elettroni di valenza del metallo da leganti carichi negativamente con una distribuzione uniforme di carica negativa attorno al metallo (campo elettrico sfericamente simmetrico), l'energia di tutti e cinque D gli orbitali aumenteranno della quantità E 0 rispetto a M libero n+ ione. Perché il D gli orbitali hanno orientamenti spaziali diversi, quindi con la concentrazione di cariche negative sui ligandi situati sugli assi delle coordinate, l'aumento della loro energia differisce. Sferzata d'energia d z 2 e d x 2- y 2 gli orbitali diretti verso i ligandi sugli assi coordinati hanno un aumento di energia maggiore dxy, dxz e dyz orbitali diretti tra gli assi delle coordinate.
Energia di fissionequintuplo degenere ( N -1) orbitali in doppiamente degeneri d x 2- y 2, z 2 orbitali e triplamente degeneri dxy,xz,yz gli orbitali sono chiamati (Fig. 26) parametro di divisione del campo cristallino. Dall'energia della scissione D orbitali nel campo ottaedrico dei ligandi non cambia rispetto al campo elettrico a simmetria sferica, quindi l'aumento dell'energia dei due d x 2- y 2, z 2 gli orbitali si verificano a 0,6D 0 e una diminuzione dell'energia di tre orbitali d xy , xz , yz di 0,4 D 0 .
Per indicare il grado di degenerazione e simmetria degli orbitali metallici divisi sotto l'influenza del campo elettrico dei ligandi, vengono utilizzati simboli speciali. Tripla degenere e simmetrica rispetto al centro di simmetria e rotazione attorno agli assi coordinati d xy , xz , yz t 2 g ", pur essendo doppiamente degenere e anch'esso simmetrico rispetto al centro di simmetria d x 2- y 2, z 2 gli orbitali sono indicati dal simbolo " per esempio " Pertanto, sotto l'influenza del campo elettrico ottaedrico dei ligandi, quintuplicato ( n-1) d gli orbitali dell'agente complessante sono divisi in orbitali triplamente e doppiamente degeneri di diversa energia t 2 orbitali g ed eg.
Una considerazione simile della variazione di energia di quintuplo degenere ( n-1) d orbitali di uno ione metallico libero in un ambiente tetraedrico di ligandi in [ ML 4] z complessi mostra (Fig. 27) la loro suddivisione anche in duplice (e) e triplice ( T ) orbitali degeneri, tuttavia, con la posizione energetica opposta. Pedice " G " quando indicato con "e" e " T » gli orbitali non sono indicati poiché il complesso tetraedrico non ha un centro di simmetria. Una diminuzione del numero di ligandi di un complesso tetraedrico rispetto a un complesso ottaedrico porta ad una diminuzione naturale del parametro di scissione del campo cristallino:D T = 4/9 D DI .
Riducendo la simmetria dell'ambiente del ligando del metallo, ad esempio, la distorsione tetragonale dell'ottaedro [ ML 6] z complessi associati all'estensione dei legami metallo-ligando con ligandi assiali [ ML 4 X 2] z e la formazione nel caso limite di piano-quadrato [ ML 4] z complessi, porta (Fig. 28) a un'ulteriore suddivisione della valenza ( n-1) d orbitali metallici.
Riempimento della divisione ( n-1) d orbitali metallici avviene secondo i principi di Pauli e l'energia minima. Per complessi ottaedrici con d1, d2 e d3 configurazione elettronica del metallo, gli elettroni di valenza, secondo la regola di Hund, si popolano t2 gr orbitali con spin paralleli, che portano a t 2 g 1 , t 2 g 2 e t 2 g 3 struttura elettronica dei complessi.
Per metalli con d 4 configurazione elettronica, si popolano anche tre elettroni t2 gr orbitali con spin paralleli. La popolazione del quarto elettrone dipende dai costi energetici per il valore dell'energia di accoppiamento degli spin (E sp.-sp.) durante la popolazione t2 gr orbitali con spin antiparallelo e violazione della regola di Hund, o superamento dell'energia di scissione da parte del campo cristallinoD o al momento del check-in, ad es orbitali con spin parallelo secondo la regola di Hund. Nel primo caso si forma un complesso con t2g4 struttura elettronica e molteplicità di spin ridotta rispetto al metallo libero 2 S+1 = 3 (S - giro totale), chiamato a basso spin. Quando la regola di Hund è soddisfatta e il quarto elettrone è popolato per esempio orbitali, si forma un complesso con t 2 g 3 e g 1 struttura elettronica e multipletto di spin libero simile al metallo 2 S +1 = 5. Tali complessi sono chiamati alta rotazione.
Allo stesso modo, quando si distribuisce la valenza d5, d6 e d7 elettroni metallici t 2 ge ad es orbitali di complessi ottadrici a seconda del rapporto E sp.-sp. ED O È possibile la formazione di due tipi di complessi:
A E sp.-sp. > D O si formano complessi ad alto spin con la struttura elettronica del metallo t 2 g 3 e g 2 , t 2 g 4 e g 2 , t 2 g 5 e g 2 secondo la regola di Hund e molteplicità di spin libera simile al metallo - 2 S +1 = 6, 5, 4;
E sp.-sp.< D O si formano complessi a basso spin con la struttura elettronica del metallo t 2 g 5 e g 0 , t 2 g 6 e g 0 , t 2 g 6 e g 1 e molteplicità di spin inferiore rispetto al metallo libero 2 S +1 = 2, 1, 2.
Complessi metallici con d8, d9 e d10 la configurazione elettronica è caratterizzata da un tipo di distribuzione degli elettroni: t2 g 6 e g 2 , t 2 g 6 e g 3 , t 2 g 6 e g 4 con molteplicità di spin simile al metallo libero: 2 S +1 = 3, 2 e 0.
Quindi il parametroD, caratterizzando la scissione ( n-1) d gli orbitali metallici sotto l'influenza del campo elettrico dei ligandi è una delle principali caratteristiche dei cambiamenti nelle proprietà dei complessi rispetto a uno ione metallico libero. È il valore del parametroDdetermina per un numero di configurazioni elettroniche del metallo determina la possibilità della formazione di complessi ad alto o basso spin con diverse distribuzioni di elettroni su orbitali divisi e diverse proprietà.
Il valore del parametro di suddivisione del campo cristallinoDdipende dalla natura del metallo dell'agente complessante, dai ligandi che lo circondano e dalla loro posizione spaziale attorno all'agente complessante:
1. Leganti in ordine di parametro crescenteDpoiché i complessi dello stesso metallo e struttura geometrica simile si trovano nelle cosiddette serie spettrochimiche: IO -< Br - < Cl - < F - < OH - < C 2 O 4 2- ~ H 2 O < NCS - < NH 3 ~ En < NO 2 - < CN - < CO . All'inizio della fila ci sono ligandi “a campo debole” - ioni alogenuro, ioni idrossido e ossalato, acqua, che formano prevalentemente complessi ad alto spin. I ligandi sul lato destro della serie: ioni monossido di carbonio, cianuro e nitrito sono chiamati ligandi “ad alto campo” e sono tipicamente caratterizzati dalla formazione di complessi a basso spin. Per i ligandi al centro della serie: ione tiocianato, ammoniaca, etilendiammina, a seconda della natura del metallo, si formano complessi ad alto o basso spin.
2. Aumentare l'efficienza del campo elettrico dei ligandi attivi D orbitali metallici con aumento della loro dimensione nella riga 3 D<< 4 d < 5 d , così come un aumento del grado di ossidazione del metallo porta ad un aumento del parametroD nella serie: Mn(II)< Ni (II ) < Co (II ) < Fe (II ) < V (II ) < Fe (III ) < Co (III ) < Mn (IV ) < Mo (III ) < Rh (III ) < Ru (III ) < Pd (IV ) < Ir (III ) < Pt (IV ).
3. Parametro Dper i complessi tetraedrici è solo 4/9 del parametroDcomplessi ottaedrici.
Complessi “pesanti” 4 d e 5 d i metalli, quasi indipendentemente dalla natura dei ligandi, formano prevalentemente complessi a basso spin, mentre la formazione di complessi a basso o alto spin è “leggera” 3 D metalli è determinata principalmente dalla forza del campo del ligando.
In contrasto con l'MMS, la teoria del campo cristallino giustifica la differenza nelle proprietà magnetiche dei complessi dello stesso ione metallico con diversi ambienti di ligando, ad esempio diamagnetici [ Fe(CN ) 6] 4- e paramagnetico [ Fe(H2O ) 6 ] 2+ non utilizza l'ipotesi della loro intraorbitale ( d2 sp3 ibridazione) e orbitale esterno ( sp 3 d 2 struttura dell'ibridazione). La differenza nelle proprietà magnetiche è determinata dalla natura a basso e ad alto spin della distribuzione degli elettroni 6-valenti Fe(II ) per suddivisione t 2 ge ad es orbitali (Fig. 29). Essendo ligandi di campo forti e deboli, si formano ioni cianuro e molecole d'acqua Fe(II ) complessi a basso e alto spin con t 2 g 6 e g 0 e t 2 g 4 e g 2 distribuzione degli elettroni, che determina il diamagnetismo [ Fe(CN ) 6 ] 4- e paramagnetismo [ Fe(H2O ) 6 ] 2+ complessi.
Divisione di quintuplo degenere ( n-1) d orbitali metallici nei complessi e cambiamenti di parametriDa seconda della natura dei leganti determina il colore caratteristico dei complessi sia allo stato solido che in soluzione. Quando il complesso assorbe la radiazione elettromagnetica nella regione visibile dello spettro (400-750) nm, l'energia dei quanti è E uguale al valore D, avviene il trasferimento di elettroni t 2 g ad es orbitali. È la radiazione elettromagnetica non assorbita della regione visibile dello spettro che determina il colore del complesso secondo il “cerchio dei colori di Newton” (Fig. 30), che mostra i colori primari e secondari della radiazione visibile.
Titanio Aquacomplex( III) [Ti (H 2 O) 6] 3+ c t 2 g 1 e g 0 distribuzione elettronica come risultato della fotoeccitazione, corrispondente alla transizione dell'elettrone ad un'energia più elevata es. orbitali:
3+ (t 2g 1 e g 0) + hN= * 3+ (t 2g 0 e g 1)
assorbe i quanti di luce nella regione gialla dello spettro, che porta al suo colore viola. Un cambiamento nell'ambiente del ligando dello ione metallico in conformità con la posizione del ligando nella serie spettrochimica porta ad un cambiamento nel parametroDe, di conseguenza, ad un cambiamento nell'energia e nella lunghezza d'onda dei quanti assorbiti dal complesso e al colore caratteristico del complesso - ad esempio, nella serie [ CuCl 4 ] 2- , [ Cu (H 2 O ) 4 ] 2+ , [ Cu (NH 3 ) 4 ] 2+ il colore dei complessi cambia da verde a blu e viola.
Insieme all'energia che divide il campo cristallinoD, svolge anche un ruolo importante in TCH energia di stabilizzazione del campo cristallino(ESKP) - guadagno di energia quando si distribuiscono gli elettroni tra quelli divisi nel complesso ( n-1) d orbitali metallici rispetto all'energia di cinque volte degenere ( n-1) d orbitali metallici in un campo elettrico sferico equivalente (Fig. 31, 32).
ESCP dei complessi ottadrali e tetraedrici.|
Mn+ |
Complessi ottaedrici |
Complessi tetraedrici |
|
|
Bassa rotazione |
Rotazione elevata |
Rotazione elevata |
|
|
0.4 D o |
0.6 D T |
||
|
0.8 D o |
1.2 D T |
||
|
1.2 D o |
0.8 D T |
||
|
d4 |
1.6 D o |
0.6 D o |
0.4 D T |
|
d5 |
2.0 D o |
0 D o |
0 D T |
|
d6 |
2.4 D o |
0.4 D o |
0.6 D T |
|
d7 |
1.8 D o |
0.8 D o |
1.2 D T |
|
d8 |
1.2 D o |
0.8 D T |
|
|
d9 |
0.6 D o |
0.4 D T |
|
|
d10 |
0 D o |
||
Una stima del valore EXP del complesso si ottiene sulla base dei diagrammi di suddivisione ( n-1) d orbitali metallici nel campo elettrico dei ligandi, mostrando una diminuzione o un aumento dell'energia del sistema rispetto a un campo elettrico sferico quando gli elettroni popolano la divisione ( n-1) d orbitali. Per ottaedrico [ ML 6] z complessi (Fig. 32) popolazione di ciascun elettrone t2 gr orbitali porta ad un guadagno di energia del sistema di 0,4D oh, check-in, ad es richiede un dispendio energetico 0,6D O . Per tetraedrico [ ML 4] z complessi con posizioni energetiche opposte e e t orbitali metallici: occupazione di ciascun elettrone per scissione e e t orbitali è accompagnato da una diminuzione e un aumento dell'energia del sistema di 0,6D t e 0,4 D T .
Essendo un riflesso della stabilità termodinamica dei complessi, le stime dei loro valori ESCR sono coerenti con i dati sperimentali sui cambiamenti nell'energia del reticolo cristallino per complessi di esafluoruro ad alto spin 3 D metalli (Fig. 33).
I valori ESC ci consentono di determinare l'isomero di coordinazione preferito (Fig. 34), ad esempio [ Cu (NH 3 ) 6 ][ NiCl 4 ] o [ Ni (NH 3 ) 6 ][ CuCl 4 ]. Per fare ciò, calcola la differenza in ESC per il catione e l'anione complessi degli isomeri. Valore ESCR [ Cu (NH 3 ) 6 ] 2+ e [NiCl 4 ] 2- è 0,6 D oe 0,8 D T rispettivamente. Considerando cheD t = 4/9 D o , la differenza tra i valori ESCP [ Cu(NH3)6]2+ e [NiCl4 ] 2- sarà il 19/45D o . Allo stesso modo, i valori di ESKP [ Ni (NH 3 ) 6 ] 2+ e [CuCl 4 ] 2- è 1,2 D oe 0.4 D T e la differenza tra loro è 28/45D o . Grande differenza catione complesso ESCP [ Ni (NH 3 ) 6 ] 2+ e l'anione [CuCl 4 ] 2- rispetto a [ Cu(NH3)6]2+ e [NiCl4 ] 2- mostra una formazione più preferibile dell'isomero della composizione [ Ni (NH 3 ) 6 ][ CuCl 4 ].
Insieme alle proprietà magnetiche e ottiche dell'influenza della struttura elettronica del metallo sulla stabilità termodinamica dei complessi, TKP prevede una distorsione della struttura geometrica dei complessi con una distribuzione non uniforme degli elettroni sulla divisione ( n-1) d orbitali metallici (Fig. 35). In contrasto con la struttura ottaedrica regolare [ Co (CN) 6] 3- s t 2 g 6 e g 0 distribuzione elettronica, distorsione tetragonale di un complesso simile [ Cu (CN) 6 ] 4- s t 2 g 6 e g 3 distribuzione elettronica contenente 3 elettroni su 2 volte degenere per esempio orbitali, porta alla trasformazione effettiva dell'ottaedrico in un complesso quadrato-planare:
4- = 2- + 2CN - .
Tutto quanto sopra mostra che la relativa semplicità e le ampie capacità del TCT per spiegare e prevedere le proprietà fisico-chimiche dei complessi determinano la grande popolarità di questo modello per descrivere i legami chimici nei composti complessi. Allo stesso tempo, concentrandosi sui cambiamenti nella struttura elettronica del metallo durante la formazione del complesso, il TCP non tiene conto della struttura elettronica dei ligandi, considerandoli come cariche puntiformi o dipoli. Ciò porta a una serie di limitazioni del TCP nella descrizione della struttura elettronica dei complessi. Ad esempio, nell'ambito del TCP è difficile spiegare la posizione di un certo numero di ligandi e metalli in serie spettrochimiche, che è associata ad un certo grado di covalenza e alla possibilità della formazione di più legami metallo-ligando. Queste limitazioni vengono eliminate se si considera la struttura elettronica di composti complessi utilizzando il metodo più complesso e meno visivo degli orbitali molecolari.
Teoria del legame di valenza è stata la prima delle teorie della meccanica quantistica utilizzata per spiegare approssimativamente la natura dei legami chimici nei composti complessi. La sua applicazione era basata sull'idea di meccanismo donatore-accettore formazione di legami covalenti tra il ligando e l'agente complessante. Legante conta particella donatrice, capace di trasferire una coppia di elettroni accettore – agente complessante, che fornisce celle quantistiche libere (orbitali atomici) dei suoi livelli energetici per la formazione di legami.
Per la formazione di legami covalenti tra l'agente complessante e i ligandi, è necessario che il vuoto S-, P- O D-gli orbitali atomici dell'agente complessante sono stati sottoposti ibridazione un certo tipo. Gli orbitali ibridi occupano una certa posizione nello spazio e il loro numero corrisponde a numero di coordinazione agente complessante.
Questo accade spesso combinando elettroni spaiati agente complessante in coppie, che consente il rilascio di un certo numero di cellule quantistiche - orbitali atomici, che poi partecipano all'ibridazione e alla formazione di legami chimici.
Le coppie solitarie di elettroni dei ligandi interagiscono con gli orbitali ibridi dell'agente complessante e sovrapposizione orbitali corrispondenti dell'agente complessante e del ligando con la comparsa di una maggiore densità elettronica nello spazio internucleare. Le coppie di elettroni dell'agente complessante, a loro volta, interagiscono con gli orbitali atomici vacanti del ligando, rafforzando la connessione attraverso il meccanismo del dativo. Pertanto, il legame chimico nei composti complessi è comune covalente connessione sufficiente durevole E energeticamente favorevole.
Le coppie di elettroni situate negli orbitali ibridi dell'agente complessante tendono ad occupare una posizione nello spazio in cui la loro mutua repulsione è minima. Questo porta a struttura ioni e molecole complessi sembrano essere in una certa dipendenza da tipo di ibridazione.
Consideriamo la formazione di alcuni complessi dal punto di vista della teoria dei legami di valenza. Innanzitutto notiamo che gli orbitali di valenza degli atomi degli agenti complessanti sono vicini in energia:
E (N- 1)D » E ns » E n.p. » E nd
|
Tipo di ibridazione |
Geometria del complesso |
||
|
lineare |
-
|
||
|
triangolare |
- |
||
|
tetraedro |
2-
|
||
|
2-
|
|||
|
sp 3 D(z 2) |
bipiramide trigonale |
||
|
sp 3 D(X 2 - sì 2) |
piramide quadrata |
3-
|
|
|
sp 3 D 2 , |
3+ |
||
|
sp 3 D 3 |
bipiramide pentagonale |
4-
|
Ad esempio, il catione 2+ comprende l'agente complessante zinco(II). Il guscio elettronico di questo ione convenzionale ha la formula 3 D 10 4S 0 4P 0 e può essere convenzionalmente rappresentato come segue:
Vacante 4 S- e 4 P-gli orbitali dell'atomo di zinco(II) formano quattro sp Orbitali 3-ibridi orientati verso i vertici del tetraedro.
Ogni molecola di ammoniaca ha una coppia solitaria di elettroni sull'atomo di azoto. Gli orbitali degli atomi di azoto contenenti coppie solitarie di elettroni si sovrappongono sp Orbitali 3-ibridi di zinco (II), che formano un catione complesso tetraedrico di tetraammina zinco (II) 2+: 
Poiché lo ione 2+ non ha elettroni spaiati, si esibisce diamagnetico proprietà.
Lo ione tetracloromanganato (II) 2- contiene cinque elettroni spaiati per 3 D-orbitali e vacanti 4 S- e 4 P-orbitali. Forma degli orbitali vacanti sp Orbitali 3-ibridi che si sovrappongono P-orbitali atomici degli ioni cloruro: 
Lo ione tetraedrico 2- così ottenuto è paramagnetico, poiché contiene cinque elettroni spaiati.
Utilizzando un algoritmo di previsione convenzionale tipo di ibridazione degli orbitali atomici nell'ambito del metodo del legame di valenza, è possibile determinare geometria dei complessi di diversa composizione. Per fare ciò, prima di tutto, è necessario scrivere la formula elettronica per il livello di valenza e costruire un diagramma della distribuzione degli elettroni sulle celle quantistiche. Ad esempio, per un atomo di nichel neutro:

Transizione 4 S-elettroni per 3 D-trasformazioni di sottolivello paramagnetico Atomo di Ni 0 pollici diamagnetico particella Ni*: 
Gli orbitali vacanti risultanti subiscono ibridazione, formando una configurazione tetraedrica. Costruito così tetraedrico diamagnetico complesso tetracarbonilnichel (CN = 4), caratterizzato da una significativa stabilità.
Se l'agente complessante è nichel (II) con configurazione elettronica 3 D 8 4S 0 4P 0, quindi la necessità di spostare gli elettroni da 4 S-sottolivello prima che l'ibridazione scompaia, poiché esiste un numero sufficiente di orbitali liberi per realizzare la coordinazione numero 4: 
Questa struttura ha un instabile paramagnetico complesso tetrabromonicolato(II)-ione 2-. Tuttavia, quando si combinano due elettroni 3 D-sottolivello in una coppia e la trasformazione di una delle celle quantistiche di questo sottolivello in una cella vacante cambia sia il tipo di ibridazione che le caratteristiche del complesso risultante: 
Tipo di ibridazione sp 2 e la forma plano-quadrata del complesso si realizzano sulla formazione di una stalla diamagnetico complesso tetracianonicolato(II)-ione 2- (CN = 4): 
Se la sintesi del complesso cianuro viene effettuata in condizioni di eccesso di ligando, si può realizzare un numero di coordinazione pari a 5: 
Stabile diamagnetico il complesso pentacianonicolato(II)-ione 3- ha la forma di una piramide quadrata: 
Complesso ottaedrico di nichel (II) 2+, sebbene paramagnetico, ma abbastanza stabile. La sua educazione è dovuta sp 3 D 2 -ibridazione degli orbitali atomici del nichel: 
Se gli orbitali atomici dell'esterno D-sottolivello, complesso, di regola, in larga misura paramagnetico e viene chiamato orbitale esterno O alta rotazione. La struttura di tali complessi può corrispondere al tipo di ibridazione, ad esempio, sp 3 D 2 .
Tali complessi, durante la formazione dei quali avviene l'ibridazione con la partecipazione degli orbitali atomici dell'esterno D-vengono chiamati i sottolivelli intraorbitale O a basso spin e, di regola, diamagnetico O debolmente paramagnetico(tutti o quasi tutti gli elettroni dell'agente complessante sono accoppiati e il tipo di ibridazione, ad es. D 2 sp 3 o sp 2).
Quando si esaminano i complessi del ferro (II), si trovano sia i complessi orbitali esterni che quelli intraorbitari.


Lo schema seguente mostra come si formano paramagnetico ad alto spin esafluoroferrato (II)-ione 4- e diamagnetico a basso spin ione esacianoferrato (II) 4-.
La stessa teoria dei legami di valenza non risponde alla domanda su quale tipo di complesso si forma in ciascun caso specifico, poiché questo metodo non tiene conto dell'influenza della natura del ligando. Pertanto, il metodo del legame di valenza deve necessariamente essere integrato con dati sulle proprietà magnetiche del complesso o informazioni sull'influenza del ligando sulla natura del complesso formato.
.Teoria del campo cristallino sostituì la teoria dei legami di valenza negli anni '40 del XX secolo. Nella sua forma pura non è attualmente utilizzata, poiché non può spiegare la formazione di legami covalenti in composti complessi e non tiene conto del vero stato dei ligandi (ad esempio, delle loro dimensioni effettive) anche nel caso di interazioni strette a puramente elettrostatico.
Già a metà degli anni '50 la teoria semplificata del campo cristallino fu sostituita da una teoria migliorata teoria dei campi dei ligandi, tenendo conto della natura covalente dei legami chimici tra l'agente complessante e il legante.
Tuttavia, l'approccio più generale per spiegare la formazione di composti complessi è dato da teoria degli orbitali molecolari(MO), che attualmente prevale su tutte le altre. Il metodo degli orbitali molecolari prevede sia l'interazione puramente elettrostatica in assenza di orbitali atomici sovrapposti, sia l'intero insieme di gradi intermedi di sovrapposizione.
Diamo un'occhiata ai concetti di base teoria del campo cristallino, che, come la teoria dei legami di valenza, conserva ancora la sua importanza per la descrizione qualitativa dei legami chimici nei composti complessi grazie alla sua grande semplicità e chiarezza.
Nella teoria del campo cristallino viene considerato il legame chimico tra l'agente complessante e il ligando elettrostatico. Secondo questa teoria i ligandi si trovano attorno all’agente complessante ai vertici dei poliedri regolari ( poliedri) COME cariche puntuali. La teoria non tiene conto del volume effettivo del ligando.
I ligandi, come le cariche puntiformi, si creano attorno all'agente complessante campo elettrostatico(“campo cristallino”, se consideriamo un cristallo di un composto complesso, oppure campo dei ligandi), in cui i livelli energetici dell'agente complessante e, soprattutto, D-sottolivelli si stanno dividendo e la loro energia cambia. Dalla natura della scissione dipende l'energia dei nuovi livelli energetici simmetria disposizione dei ligandi (ottaedrico, tetraedrico o altro campo cristallino). Quando le molecole H 2 O, NH 3 , CO e altre sono coordinate come ligandi, sono considerate come dipoli, orientato con carica negativa verso l'agente complessante.
Consideriamo il caso di una disposizione ottaedrica di ligandi (ad esempio 3- o 3+). Al centro dell'ottaedro c'è un atomo complessante M(+n) con elettroni accesi D-orbitali atomici, e ai suoi vertici ci sono ligandi sotto forma di cariche puntiformi negative (ad esempio ioni F o molecole polari come NH 3). In uno ione convenzionale M(+n) non associato a ligandi, le energie di tutti e cinque D-AO sono gli stessi (cioè gli orbitali atomici degenerare).
Tuttavia, nel campo ottaedrico dei ligandi D-AO dell'agente complessante rientrano disuguale posizione. Orbitali atomici D(z 2) e D(X 2 -
sì 2), allungate lungo gli assi coordinati, si avvicinano di più ai leganti. Tra questi orbitali e i ligandi situati ai vertici dell'ottaedro sorgono differenze significative forze repulsive, portando ad un aumento dell'energia orbitale. In altre parole, questi orbitali atomici sono soggetti a massima esposizione al campo del ligando. Una molla fortemente compressa può servire da modello fisico di tale interazione. 

Altri tre D-AO- D(xy), D(xz) E D(sì), posti tra gli assi coordinati e tra i leganti, sono ad una distanza maggiore da essi. L'interazione di tale D-AO con i ligandi è minimo e quindi energetico D(xy), D(xz) E D(sì)-AO diminuisce rispetto a quello originale. 

Quindi, quintuplicato degenere D-AO agente complessante, entrando campo del legante ottaedrico, esposto scissione in due gruppi di nuovi orbitali – orbitali triplamente degeneri con energia minore, D(xy), D(xz) E D(sì), E orbitali doppiamente degeneri con energia più elevata D(z 2) e D(X 2 -
sì 2). Questi nuovi gruppi D-orbitali con inferiore E energia più elevata denota D e e D G: 
Differenza energetica due nuovi sottolivelli D e e D g ho il nome parametro di suddivisione D0:
E 2 – E 1 = D0
Posizione di due nuovi sottolivelli energetici D e e D g rispetto all'originale ( D-AO) sul diagramma energetico asimmetrico:
(E 2 – E 0) > (E 0 – E 1).
Teoria quantistica lo richiede quando i nuovi livelli energetici sono completamente popolati di elettroni, l'energia totale rimane invariata, cioè. dovrebbe restare uguale a E 0 .
In altre parole, l’uguaglianza deve essere soddisfatta
4(E 2 – E 0) = 6(E 0 – E 1),
dove 4 e 6 – massimo numero di elettroni per D g - e D e-AO. Da questa uguaglianza ne consegue che
(E 2 – E 0) / (E 0 – E 1) = 3/2 e
(E 2 – E 1) / (E 0 – E 1 >) = 5/2, o
D0/( E 0 – E 1) = 5/2, da cui ( E 0 – E 1) = 2/5 ´ D 0 >. Posizionando ogni elettrone tra i sei possibili su D Cause degli orbitali elettronici diminuire (vincite) energia per 2/5 D 0 . Al contrario, il posizionamento di ciascun elettrone su quattro è possibile D Causa degli orbitali g aumento (costo) energia per 3/5 D 0 . Se popolato di elettroni D e - e D g -orbitali completamente, quindi no vincente energia non lo farà(proprio come non accadrà consumo energetico aggiuntivo): 4 ´ 3/5 ´ D 0 - 6 ´ 2/5 ´ D 0 = 0. Ma se l'originale D-AO è popolato solo parzialmente e contiene da 1 a 6 elettroni, e questi elettroni sono posizionati solo su D e -AO, allora otteniamo notevole guadagno energetico. La specificità di ciascun ligando influenza il campo creato da questo ligando: forte O Debole. Come campo più forte ligandi rispetto a Di più Senso parametro di suddivisione D0. Lo studio del parametro di scissione è solitamente basato su spettroscopico ricerca. Lunghezze d'onda bande di assorbimento complessi l allo stato cristallino o in soluzione, a causa della transizione di elettroni da D e - su D g-AO, associato a parametro di suddivisione D0 come segue: n = 1/l; D dove è la costante di Planck H pari a 6.626 ´ 10 - 34 J. s; Parametro di divisione, oltre al tipo di ligando, dipende dal grado di ossidazione E natura agente complessante. A aumento della carica nucleare aumenta anche l'atomo D 0 che forma il complesso. Cationi esaamminiridio(III) 3+, esaamminiridio(III) 3+ e esaamminiridio(III) 3+ ( Z= 27, 45 e 77) sono caratterizzati da parametri di frazionamento pari a 22900, 34100 e 41000 cm -1. La dipendenza di D0 dalla natura dei ligandi è più diversificata. Come risultato dello studio di numerosi composti complessi, si è scoperto che in termini della loro capacità di aumentare il parametro di scissione dei metalli complessanti situati nei loro consueti stati di ossidazione, i ligandi più comuni possono essere organizzati nei seguenti serie spettrochimiche, lungo il quale il valore di D 0 aumenta monotonicamente: Pertanto, il campo elettrostatico attorno all'agente complessante è più forte e la scissione più forte D-AO è causato dai ligandi NO2, CN -
e CO. Consideriamo la distribuzione degli elettroni su D e - e D Orbitali g nel campo ottaedrico dei ligandi. Registrare D e - e D gli orbitali g si verificano in pieno accordo con La regola di Hund E Principio di Pauli. In questo caso, indipendentemente dal valore del parametro di splitting, i primi tre elettroni sono occupati dalle celle quantistiche D sottolivello e: Se il numero di elettroni per D- i sottolivelli dell'agente complessante sono più di tre; vi sono due possibilità per collocarli su sottolivelli suddivisi. Ad un valore basso del parametro di split (campo debole dei ligandi), gli elettroni superano la barriera energetica che li separa D e - e D orbitali g; il quarto e poi il quinto elettrone popolano le cellule quantistiche D sottolivello g. Con un forte campo del ligando e un elevato valore D0, la popolazione è popolata dal quarto e quinto elettrone D escluso il sottolivello g; compilazione in corso D orbitali elettronici. A ligandi di campo deboli popolando celle quantistiche con 4 o 5 elettroni giri paralleli, quindi il complesso risultante risulta essere fortemente paramagnetico. In un forte campo di ligandi si formano una e poi due coppie di elettroni D e -sottolivello, quindi paramagnetismo il complesso risulta essere molto più debole. Il sesto, il settimo e l'ottavo elettrone in caso di campo debole si riaccendono D e -sottolivello, che completa le configurazioni delle coppie di elettroni (uno nel caso D 6, due – D 7 e tre - D 8): Nel caso di un forte campo del ligando, si popola il sesto elettrone D e -AO, che porta a diamagnetismo complesso, dopo di che vanno il settimo e l'ottavo elettrone D sottolivello g: Ovviamente, con una configurazione a otto elettroni differenze di struttura tra complessi con ligandi Debole E i campi forti scompaiono. Anche l'occupazione degli orbitali da parte del nono e del decimo elettrone non differisce per i complessi di entrambi i tipi: Torniamo alla considerazione della struttura elettronica degli ioni complessi ottaedrici 3+ e 3-. Secondo la posizione in serie spettrochimiche, l'ammoniaca NH 3 è uno dei ligandi campo forte
, e ione fluoruro F - – campo debole
. Di conseguenza, l'occupazione degli orbitali atomici da parte degli elettroni in questi complessi avverrà secondo il seguente schema: Nell'anione 3-, i ligandi F- creano un campo cristallino debole (D 0 = 13000 cm - 1) e tutti gli elettroni dell'anione 3 D 6 -JSC si trovano su D e - e D orbitali g senza alcun accoppiamento. Uno ione complesso lo è alta rotazione e contiene quattro elettroni spaiati, quindi paramagnetico. Nello ione 3+, i ligandi NH 3 creano un forte campo cristallino (D 0 = 22900 cm - 1), tutti e 3 D 6 elettroni sono posizionati in una posizione energeticamente più favorevole D orbitali elettronici. Trasferimento di elettroni da D e - su D orbitali g impossibile anche perché barriera ad alta energia. Pertanto, questo catione complesso è rotazione bassa, non contiene elettroni spaiati e diamagnetico. In modo simile, possono essere presentati schemi per la distribuzione degli elettroni sugli orbitali in un campo ottaedrico per ioni 2+ e 4-: I ligandi H 2 O creano un campo debole; scambio di elettroni tra D e - e D Gli orbitali g non causano alcuna difficoltà e quindi il numero di elettroni spaiati nello ione complesso è lo stesso dello ione Fe + II convenzionale. Il complesso acquatico risultante è ad alto spin, paramagnetico. Molti composti complessi allo stato cristallino e in soluzione acquosa sono colorati brillantemente. Pertanto, una soluzione acquosa contenente 2+ cationi è colorata intensamente di blu, 3+ cationi danno alla soluzione un colore viola e 2+ cationi le danno un colore rosso. La teoria del campo cristallino permette di spiegare l'aspetto di un colore o di un altro nei composti complessi. Se la luce viene fatta passare attraverso una soluzione o un campione cristallino di una sostanza parte visibile dello spettro, allora, in linea di principio, sono possibili tre opzioni per il comportamento fisico del campione: nessun assorbimento della luce qualsiasi lunghezza d'onda (campione di sostanza incolore, sebbene possa avere bande di assorbimento nella regione ultravioletta dello spettro); completo assorbimento della luce sull'intero intervallo di lunghezze d'onda (il campione apparirà nero); Finalmente, assorbimento della luce soltanto determinata lunghezza d'onda(quindi il campione avrà colore complementare a quello assorbito parte ristretta dello spettro). Pertanto, viene determinato il colore della soluzione o dei cristalli frequenza delle bande di assorbimento luce visibile: L'assorbimento dei quanti di luce da parte dei complessi (ad esempio quelli con struttura ottaedrica) è spiegato dall'interazione della luce con gli elettroni situati su D e -sottolivello, accompagnato dalla loro transizione verso orbitali vacanti D sottolivello g. Ad esempio, quando si fa passare la luce attraverso una soluzione acquosa contenente cationi esaacquatitanio(III) 3+, viene rilevata una banda di assorbimento della luce nella regione giallo-verde dello spettro (20300 cm - 1, l » 500 nm). Ciò è dovuto alla transizione del singolo elettrone dell'agente complessante da D e-AO acceso D sottolivello g: Pertanto, una soluzione contenente 3+ acquisisce un colore viola (oltre al giallo-verde assorbito). Una soluzione di sale di vanadio Cl 3 è verde. Ciò è dovuto anche alle corrispondenti transizioni degli elettroni quando assorbono parte dell'energia del fascio luminoso. Nello stato fondamentale, con la configurazione elettronica del vanadio (III) 3 D 2, sono occupati due elettroni spaiati D sottolivello e: C'è solo due opzioni per la transizione di due elettroni SU D g -sottolivello: entrambi Entrambi gli elettroni sono occupati D g -AO, o solo uno di loro. Qualsiasi altra transizione elettronica associata ad una diminuzione dello spin totale è vietata. Se l'agente complessante ha una configurazione elettronica D 0 o D 10 allora transizioni elettroniche Con D e - su D g -sottolivello o viceversa impossibile o perché assenza di elettroni, o perché assenza di orbitali liberi. Pertanto, soluzioni di complessi con agenti complessanti come Sc(III), Cu(I), Zn(II), Cd(II), ecc., non assorbono energia nella parte visibile dello spettro e appaiono incolore: La selettività dell'assorbimento della luce non dipende solo da agente complessante E il suo stato di ossidazione, ma anche da tipo di ligandi. Quando si sostituiscono i ligandi sul lato sinistro della serie spettrochimica in un composto complesso con ligandi che creano forte campo elettrostatico osservato aumento la frazione di energia assorbita dagli elettroni dalla luce trasmessa e, di conseguenza, diminuire lunghezza d'onda della corrispondente banda di assorbimento. Pertanto, una soluzione acquosa contenente cationi tetraacquarame(II) 2+ è blu e una soluzione di tetraamminorame(II) 2+ solfato è intensamente blu. ________________________ Ripeto: >>> Applicazioni
Guadagno di energia dovuto a soluzione prioritaria elettroni D vengono chiamati gli orbitali atomici e energia di stabilizzazione del complesso da parte del campo del ligando.
velocità della luce Con
= 3 ´ 10 10 cm/s.
Unità D 0 è uguale a quello del numero d'onda n: cm - 1, che corrisponde approssimativamente a 12 J/mol.
Nei composti complessi che includono agenti complessanti dello stesso periodo e nello stesso stato di ossidazione, con gli stessi ligandi, il parametro di scissione è approssimativamente lo stesso. All'aumentare del grado di ossidazione dell'agente complessante, il valore di D 0 aumenta. Pertanto, per gli acquacomplessi 2+ e 2+, il valore del parametro di divisione è 7800 e 10400 cm - 1, e per 3+ e 3+ - 13700 e 21000 cm - 1, rispettivamente.
I-Br -Cl - » NCS - NUMERO 3 -F -OH -H2O »H-NH3 NO2 -CN - "NO" CO. 









Al contrario, i ligandi CN provocano una scissione significativa D-AO, pari a 33000 cm - 1. Ciò significa che c'è un forte tendenza ad allocare tutti gli elettroni SU D orbitali elettronici. Guadagno di energia, ottenuto con una tale popolazione di orbitali, è molto maggiore del costo energetico dovuto all'accoppiamento degli elettroni.

Le transizioni indicate degli elettroni che hanno ricevuto energia in eccesso corrispondono a banda di assorbimento circa 400 nm nello spettro di assorbimento di una soluzione di cloruro di esaacquavanadio(III). L'assorbimento della regione viola-viola dello spettro conferisce un colore aggiuntivo alla soluzione - verde acceso.

E John Van Vleck per descrivere gli stati inferiori dei cationi dei metalli di transizione circondati da ligandi - sia anioni che molecole neutre. La teoria del campo cristallino è stata ulteriormente combinata [e perfezionata] con la teoria degli orbitali molecolari (delocalizzati) in una teoria più generale che tiene conto della covalenza parziale del legame metallo-ligando nei composti di coordinazione.
La teoria del campo cristallino consente di prevedere o interpretare gli spettri di assorbimento ottico e di risonanza paramagnetica elettronica di cristalli e composti complessi, nonché le entalpie di idratazione e stabilità in soluzioni di complessi di metalli di transizione.
Revisione della teoria del campo cristallino[ | ]
Secondo TCP, l’interazione tra un metallo di transizione e i ligandi nasce dall’attrazione tra il catione metallico caricato positivamente e la carica negativa degli elettroni negli orbitali di non legame del ligando. La teoria considera la variazione di energia di cinque degeneri D-orbitali circondati da cariche puntiformi di ligandi. Quando il ligando si avvicina allo ione metallico, gli elettroni del ligando si avvicinano ad alcuni D-orbitali rispetto ad altri, causando una perdita di degenerazione. Elettroni D-orbitali e ligandi si respingono come cariche dello stesso segno. Quindi, l'energia di quelli D-gli elettroni più vicini ai ligandi diventano più alti di quelli più lontani, il che porta ad una suddivisione dei livelli energetici D-orbitali.
I seguenti fattori influenzano la suddivisione:
- Natura dello ione metallico.
- Il grado di ossidazione del metallo. Maggiore è lo stato di ossidazione, maggiore è l’energia di scissione.
- Disposizione dei ligandi attorno ad uno ione metallico.
- La natura dei ligandi che circondano lo ione metallico. Più forte è l'effetto dei ligandi, maggiore è la differenza tra livelli energetici alti e bassi.
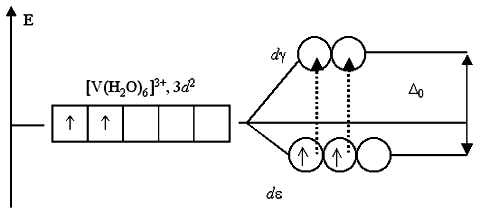
Il tipo più comune di coordinazione dei ligandi è ottaedrico, in cui sei ligandi creano un campo cristallino di simmetria ottaedrica attorno allo ione metallico. Nell'ambiente ottaedrico di uno ione metallico con un elettrone nel guscio esterno, gli orbitali d sono divisi in due gruppi con una differenza nei livelli di energia Δ oct ( energia di fissione), mentre l'energia degli orbitali dxy, dxz E d yz sarà inferiore a D z 2 e D X 2 -sì 2, poiché gli orbitali del primo gruppo si trovano più lontani dai ligandi e subiscono meno repulsione. I tre orbitali a bassa energia sono designati come t2g e due con alto - simile per esempio.
I successivi più comuni sono tetraedrico complessi in cui quattro ligandi formano un tetraedro attorno a uno ione metallico. In questo caso D-gli orbitali sono anche divisi in due gruppi con una differenza nei livelli di energia Δ tetr. A differenza della coordinazione ottaedrica, gli orbitali avranno una bassa energia D z 2 e D X 2 -sì 2, e alto - D xy , D xz E D sì. Inoltre, poiché gli elettroni dei ligandi non sono direttamente nella direzione D-orbitali, l'energia di scissione sarà inferiore rispetto alla coordinazione ottaedrica. Usando TCP puoi anche descrivere piano-quadrato e altre geometrie di complessi.
La differenza nei livelli energetici Δ tra due o più gruppi di orbitali dipende anche dalla natura dei ligandi. Alcuni ligandi causano una scissione minore di altri, le cui ragioni vengono spiegate. Serie spettrochimiche- un elenco di ligandi ottenuto sperimentalmente, ordinato in ordine crescente Δ:
Anche lo stato di ossidazione del metallo influisce su Δ. Un metallo con uno stato di ossidazione più elevato attrae i ligandi più vicini a causa di una maggiore differenza di carica. I ligandi più vicini allo ione metallico provocano una maggiore scissione.
Complessi a basso ed alto spin[ | ]
Leganti che causano una scissione maggiore D-livelli, come CN− e CO, sono chiamati ligandi campo forte. Nei complessi con tali ligandi, è sfavorevole che gli elettroni occupino orbitali ad alta energia. Di conseguenza, gli orbitali a bassa energia vengono completamente riempiti prima che gli orbitali ad alta energia inizino a riempirsi. Tali complessi sono chiamati a basso spin. Ad esempio, NO 2 − è un ligando ad alto campo che produce un ampio splitting. Tutti e 5 D-gli elettroni dello ione ottaedrico 3− saranno posizionati al livello inferiore T 2G .
Al contrario, i ligandi che causano una piccola scissione, come I− e Br−, sono chiamati ligandi campo debole. In questo caso, è più semplice posizionare gli elettroni in orbitali ad alta energia piuttosto che posizionare due elettroni nello stesso orbitale a bassa energia, perché due elettroni nello stesso orbitale si respingono a vicenda e il costo energetico per posizionare un secondo elettrone nell’orbitale è superiore a Δ. Quindi, prima che appaiano gli elettroni accoppiati, in ciascuno dei cinque D-Gli orbitali devono essere posizionati un elettrone alla volta secondo la regola di Hund. Tali complessi sono chiamati alta rotazione. Ad esempio, Br− è un ligando a campo debole che causa una piccola scissione. Tutti e 5 D-orbitali dello ione 3−, che ne ha anche 5 D-gli elettroni saranno occupati da un elettrone.
L'energia di scissione per i complessi tetraedrici Δ tetr è approssimativamente uguale a 4/9 Δ oct (per lo stesso metallo e ligandi). Di conseguenza, la differenza nei livelli di energia D-gli orbitali sono solitamente al di sotto dell'energia di accoppiamento elettronico e i complessi tetraedrici hanno solitamente uno spin elevato.
Diagrammi di distribuzione D-gli elettroni consentono di prevedere le proprietà magnetiche dei composti di coordinazione. I complessi con elettroni spaiati sono paramagnetici e sono attratti da un campo magnetico, mentre i complessi senza sono diamagnetici e si respingono debolmente.
Energia di stabilizzazione del campo cristallino[ | ]
L'energia di stabilizzazione del campo cristallino (CFE) è l'energia della configurazione elettronica di uno ione di metallo di transizione rispetto all'energia media degli orbitali. La stabilizzazione avviene a causa del fatto che nel campo del ligando il livello energetico di alcuni orbitali è inferiore rispetto a un ipotetico campo sferico, in cui tutti e cinque D-gli orbitali hanno la stessa forza repulsiva, e tutto il resto D-gli orbitali sono degenerati. Ad esempio, nel caso ottaedrico il livello t2g inferiore al livello medio in un campo sferico. Di conseguenza, se questi orbitali contengono elettroni, allora lo ione metallico è più stabile nel campo del legante rispetto al campo sferico. Al contrario, il livello energetico degli orbitali per esempio sopra la media e gli elettroni in essi contenuti riducono la stabilizzazione.
Energia di stabilizzazione mediante il campo ottaedrico
Ci sono tre orbitali in un campo ottaedrico t2g stabilizzato rispetto al livello energetico medio di 2/5 Δ ottava e i due orbitali per esempio destabilizzato di 3/5 Δ ott. Sopra c'erano esempi di due configurazioni elettroniche D 5 . Nel primo esempio, c'è un complesso 3− a basso spin con cinque elettroni t2g. La sua ESP è 5 × 2 / 5 Δ oct = 2 Δ oct. Nel secondo esempio, un complesso ad alto spin 3− con ESKP (3 × 2 / 5 Δ oct) − (2 × 3 / 5 Δ oct) = 0. In questo caso, l'effetto stabilizzante degli elettroni negli orbitali di basso livello viene neutralizzato dall'effetto destabilizzante degli elettroni negli orbitali di alto livello.
Diagrammi di suddivisione del livello D da parte di un campo cristallino[ | ]
| ottaedrico | pentagonale-bipiramidale | quadrato-antiprismatico |
|---|---|---|
Teoria del campo cristallino sostituì la teoria dei legami di valenza negli anni '40 del XX secolo. Nella sua forma pura non è attualmente utilizzata, poiché non può spiegare la formazione di legami covalenti in composti complessi e non tiene conto del vero stato dei ligandi (ad esempio, delle loro dimensioni effettive) anche nel caso di interazioni strette a puramente elettrostatico.
Già a metà degli anni '50 la teoria semplificata del campo cristallino fu sostituita da una teoria migliorata teoria dei campi dei ligandi, tenendo conto della natura covalente dei legami chimici tra l'agente complessante e il legante.
Tuttavia, l'approccio più generale per spiegare la formazione di composti complessi è dato da teoria degli orbitali molecolari(MO), che attualmente prevale su tutte le altre. Il metodo degli orbitali molecolari prevede sia l'interazione puramente elettrostatica in assenza di orbitali atomici sovrapposti, sia l'intero insieme di gradi intermedi di sovrapposizione.
Diamo un'occhiata ai concetti di base teoria del campo cristallino, che, come la teoria dei legami di valenza, conserva ancora la sua importanza per la descrizione qualitativa dei legami chimici nei composti complessi grazie alla sua grande semplicità e chiarezza.
Nella teoria del campo cristallino viene considerato il legame chimico tra l'agente complessante e il ligando elettrostatico. Secondo questa teoria i ligandi si trovano attorno all’agente complessante ai vertici dei poliedri regolari ( poliedri) COME cariche puntuali. La teoria non tiene conto del volume effettivo del ligando.
I ligandi, come le cariche puntiformi, si creano attorno all'agente complessante campo elettrostatico(“campo cristallino”, se consideriamo un cristallo di un composto complesso, oppure campo dei ligandi), in cui i livelli energetici dell'agente complessante e, soprattutto, D-sottolivelli si stanno dividendo e la loro energia cambia. Dalla natura della scissione dipende l'energia dei nuovi livelli energetici simmetria disposizione dei ligandi (ottaedrico, tetraedrico o altro campo cristallino). Quando le molecole H 2 O, NH 3 , CO e altre sono coordinate come ligandi, sono considerate come dipoli, orientato con carica negativa verso l'agente complessante.
Considera il caso di una disposizione ottaedrica di ligandi (ad esempio, -3 o 3+). Al centro dell'ottaedro c'è uno ione complessante M(+ n) con elettroni accesi D-orbitali atomici, e ai suoi vertici ci sono ligandi sotto forma di cariche puntiformi negative (ad esempio ioni F o molecole polari come NH 3). In uno ione convenzionale M(+ n), non associato a ligandi, le energie di tutti e cinque D-AO sono gli stessi (cioè gli orbitali atomici degenerare).
Tuttavia, nel campo ottaedrico dei ligandi D-AO dell'agente complessante rientrano disuguale posizione. Orbitali atomici D(z 2) e d(x2 -y2), allungati lungo gli assi delle coordinate, si avvicinano di più ai leganti. Tra questi orbitali e i ligandi situati ai vertici dell'ottaedro sorgono differenze significative forze repulsive, portando ad un aumento dell'energia orbitale. In altre parole, questi orbitali atomici sono soggetti a massima esposizione al campo del ligando. Una molla fortemente compressa può servire da modello fisico di tale interazione.
Altri tre D-AO- D(xy), D(xz) E D(sì), posti tra gli assi coordinati e tra i leganti, sono ad una distanza maggiore da essi. L'interazione di tale D-AO con i ligandi è minimo e quindi energetico D(xy), D(xz) E D(sì)-AO diminuisce rispetto a quello originale.
Quindi, quintuplicato degenere D-AO agente complessante, entrando campo del legante ottaedrico, esposto scissione in due gruppi di nuovi orbitali – orbitali triplamente degeneri con energia minore, D(xy), D(xz) E D(sì), E orbitali doppiamente degeneri con energia più elevata D(z 2) e d(x2 -y2). Questi nuovi gruppi D-orbitali con inferiore E energia più elevata denota Dε e Dγ:
D(z 2) e d(x2 -y2)
D(xy), D(xz),D(sì)
Differenza energetica due nuovi sottolivelli Dε e Dè stato nominato γ parametro di suddivisione Δ 0:
E 2 – E 1 = Δ0 ≈ 0
Posizione di due nuovi sottolivelli energetici Dε e Dγ rispetto all'originale ( D-AO) sul diagramma energetico asimmetrico:
(E 2 – E 0) > (E 0 – E 1).
Teoria quantistica lo richiede quando i nuovi livelli energetici sono completamente popolati di elettroni, l'energia totale rimane invariata, cioè. dovrebbe restare uguale a E 0 .
In altre parole, l’uguaglianza deve essere soddisfatta
4(E 2 – E 0) = 6(E 0 – E 1),
dove 4 e 6 – massimo numero di elettroni per Dγ- e Dε-AO. Da questa uguaglianza ne consegue che
(E 2 – E 0) / (E 0 – E 1) = 3/2 e
(E 2 – E 1) / (E 0 – E 1) = 5/2, o
Δ 0 / ( E 0 – E 1) = 5/2, da cui ( E 0 – E 1) = 2/5Δ0 .
Posizionando ogni elettrone tra i sei possibili su D Causa degli orbitali ε diminuire (vincite) energia di 2/5 Δ 0 .
Al contrario, il posizionamento di ciascun elettrone su quattro è possibile D Causa degli orbitali γ aumento (costo) energia di 3/5 Δ 0 .
Se popolato di elettroni Dε- e D completamente gli orbitali γ, allora no vincente energia non lo farà(proprio come non accadrà consumo energetico aggiuntivo).
Ma se l'originale D-AO è popolato solo parzialmente e contiene da 1 a 6 elettroni, e questi elettroni sono posizionati solo su Dε-AO, allora otteniamo notevole guadagno energetico.
Guadagno di energia dovuto a soluzione prioritaria elettroni D vengono chiamati gli orbitali ε-atomici energia di stabilizzazione del complesso da parte del campo del ligando.
La specificità di ciascun ligando influenza il campo creato da questo ligando: forte O Debole. Come campo più forte ligandi rispetto a Di più Senso parametro di suddivisione Δ 0 .
Lo studio del parametro di scissione è solitamente basato su spettroscopico ricerca. Lunghezze d'onda bande di assorbimento complessi allo stato cristallino o in soluzione, a causa del trasferimento di elettroni da Dε- acceso Dγ-AO, associato a parametro di suddivisioneΔ 0 come segue:
λ = C / ν; Δ 0 = E 2 – E 1 = H ν = H · ( C / λ),
dove è la costante di Planck H pari a 6,6260693 ∙ 10 -34 J·s;
velocità della luce Con
= 3 · 10 10 cm/s.
UnitàΔ 0 è uguale al numero d'onda: cm -1, che corrisponde approssimativamente a 12 J/mol. Parametro di divisione, oltre al tipo di ligando, dipende dal grado di ossidazione E natura agente complessante.
Nei composti complessi che includono agenti complessanti dello stesso periodo e nello stesso stato di ossidazione, con gli stessi ligandi, il parametro di scissione è approssimativamente lo stesso. All'aumentare del grado di ossidazione dell'agente complessante, il valore Δ 0 aumenta. Pertanto, per gli acquacomplessi 2+ e 2+ il valore del parametro di suddivisione è 7800 e 10400 cm -1, e per 3+ e +3 13700 e 21000 cm -1, rispettivamente. A aumento della carica nucleare dell'atomo che forma il complesso aumenta anche Δ 0. Cationi esaamminiridio(III) 3+, esaamminiridio(III) 3+ e esaamminiridio(III) 3+ ( Z= 27, 45 e 77) sono caratterizzati da parametri di frazionamento pari a 22900, 34100 e 41000 cm -1.
La dipendenza di Δ 0 dalla natura dei ligandi è più diversificata. Come risultato dello studio di numerosi composti complessi, si è scoperto che in termini della loro capacità di aumentare il parametro di scissione dei metalli complessanti situati nei loro consueti stati di ossidazione, i ligandi più comuni possono essere organizzati nei seguenti serie spettrochimiche, lungo il quale il valore Δ 0 aumenta monotonicamente:
I > Br > Cl > NCS - ≈ NO 3 - > F - > OH - > H 2 O > H - > NH 3 > NO 2 - > CN - > NO > CO.
Pertanto, il campo elettrostatico attorno all'agente complessante è più forte e la scissione più forte D-AO è causato dai ligandi CN-, NO e CO. Consideriamo la distribuzione degli elettroni su Dε- e D Orbitali γ nel campo ottaedrico dei ligandi. Registrare Dε- e D Gli orbitali γ si verificano in pieno accordo con La regola di Hund E Principio di Pauli. In questo caso, indipendentemente dal valore del parametro di splitting, i primi tre elettroni sono occupati dalle celle quantistiche D Sottolivello ε:
![]() dε
dε
Se il numero di elettroni per D- i sottolivelli dell'agente complessante sono più di tre; vi sono due possibilità per collocarli su sottolivelli suddivisi. Ad un valore basso del parametro di split (campo debole dei ligandi), gli elettroni superano la barriera energetica che li separa Dε- e D orbitali γ; il quarto e poi il quinto elettrone popolano le cellule quantistiche D Sottolivello γ.
![]() dε
dε
Con un forte campo del ligando e un valore elevato di Δ 0, la popolazione del quarto e quinto elettrone D Il sottolivello γ è escluso; compilazione in corso D Orbitali ε.
![]() dε
dε
A ligandi di campo deboli popolando celle quantistiche con 4 o 5 elettroni giri paralleli, quindi il complesso risultante risulta essere fortemente paramagnetico. In un forte campo di ligandi si formano una e poi due coppie di elettroni Dε-sottolivello, quindi paramagnetismo il complesso risulta essere molto più debole. Il sesto, il settimo e l'ottavo elettrone in caso di campo debole si riaccendono D Sottolivello γ, che completa le configurazioni delle coppie di elettroni (una nel caso D 6, due – D 7 e tre - D 8):
![]() dε
dε
Nel caso di un forte campo del ligando, si popola il sesto elettrone dε-AO, che porta a diamagnetismo complesso, dopo di che vanno il settimo e l'ottavo elettrone D Sottolivello γ:
![]() dε
dε
Ovviamente, con una configurazione a otto elettroni differenze di struttura tra complessi con ligandi Debole E i campi forti scompaiono. Anche l'occupazione degli orbitali da parte del nono e del decimo elettrone non differisce per i complessi di entrambi i tipi:
![]() dε
dε
Torniamo alla considerazione della struttura elettronica degli ioni complessi ottaedrici 3+ e -3. Secondo la posizione in serie spettrochimiche, l'ammoniaca NH 3 è uno dei ligandi campo forte, e ione fluoruro F - – campo debole. Nell'anione -3, i ligandi F creano un campo cristallino debole (Δ 0 = 13000 cm -1) e tutti gli elettroni dell'anione 3 originale D 6 -JSC si trovano su Dε- e D Orbitali γ senza alcun accoppiamento. Uno ione complesso lo è alta rotazione e contiene quattro elettroni spaiati, quindi paramagnetico:

Nello ione 3+, i ligandi NH 3 creano un forte campo cristallino (Δ 0 = 22900 cm -1), tutti e 3 D 6 elettroni sono posizionati in una posizione energeticamente più favorevole D Orbitali ε. Trasferimento di elettroni da Dε- acceso D orbitali γ impossibile anche perché barriera ad alta energia. Pertanto, questo catione complesso è rotazione bassa, non contiene elettroni spaiati e diamagnetico:

In modo simile, possono essere presentati schemi per la distribuzione degli elettroni sugli orbitali in un campo ottaedrico per ioni 2+ e -4:


I ligandi H 2 O creano un campo debole; scambio di elettroni tra Dε- e D Gli orbitali γ non causano alcuna difficoltà e quindi il numero di elettroni spaiati nello ione complesso è lo stesso dello ione convenzionale Fe + II. Il complesso acquatico risultante è ad alto spin, paramagnetico.
Al contrario, i ligandi CN provocano una scissione significativa D-AO, pari a 33000 cm -1. Ciò significa che c'è un forte tendenza ad allocare tutti gli elettroni SU D Orbitali ε. Guadagno di energia, ottenuto con una tale popolazione di orbitali, è molto maggiore del costo energetico dovuto all'accoppiamento degli elettroni.
Dal punto di vista del metodo del legame di valenza, l'ibridazione degli orbitali di valenza che formano un legame nell'acqua complesso comporta D-AO sottolivello esterno (4 sp 3 D 2), e in basso spin - D-Sottolivello interno JSC (3 D 2 4sp 3).
Pertanto, nei complessi ad alto spin con ligandi a campo debole, l'ibridazione avviene con la partecipazione D-AO del sottolivello esterno e basso spin con ligandi ad alto campo - D- JSC del sottolivello interno. Il numero di elettroni spaiati nel complesso può essere determinato mediante risonanza paramagnetica elettronica (EPR). Utilizzando gli strumenti di questo metodo, chiamati spettrometri EPR, vengono studiate le sostanze paramagnetiche.
La teoria del campo cristallino permette di spiegare l'aspetto di un colore o di un altro nei composti complessi. Tra i composti complessi, un numero significativo allo stato cristallino e in soluzione acquosa si distinguono per il loro colore brillante. Pertanto, una soluzione acquosa contenente 2+ cationi è colorata intensamente di blu, 3+ cationi danno alla soluzione un colore viola e 2+ cationi le danno un colore rosso. Se la luce viene fatta passare attraverso una soluzione o un campione cristallino di una sostanza parte visibile dello spettro, allora, in linea di principio, sono possibili tre opzioni per il comportamento fisico del campione: nessun assorbimento della luce qualsiasi lunghezza d'onda (campione di sostanza incolore, sebbene possa avere bande di assorbimento nella regione ultravioletta dello spettro); completo assorbimento della luce sull'intero intervallo di lunghezze d'onda (il campione apparirà nero); Finalmente, assorbimento della luce soltanto determinata lunghezza d'onda(quindi il campione avrà colore complementare a quello assorbito parte ristretta dello spettro).

Pertanto, viene determinato il colore della soluzione o dei cristalli frequenza delle bande di assorbimento luce visibile. L'assorbimento dei quanti di luce da parte dei complessi (ad esempio quelli con struttura ottaedrica) è spiegato dall'interazione della luce con gli elettroni situati su D Sottolivello ε, accompagnato dalla loro transizione verso orbitali vacanti D sottolivello γ. Ad esempio, quando si fa passare la luce attraverso una soluzione acquosa contenente cationi esaacquatitanio(III) 3+, viene rilevata una banda di assorbimento della luce nella regione giallo-verde dello spettro (20300 cm -1, λ=500 nm). Ciò è dovuto alla transizione del singolo elettrone dell'agente complessante da Dε-AO acceso D Sottolivello γ:

Pertanto, una soluzione contenente 3+ acquisisce un colore viola (oltre al giallo-verde assorbito). Una soluzione di sale di vanadio Cl 3 è verde. Ciò è dovuto anche alle corrispondenti transizioni degli elettroni quando assorbono parte dell'energia del fascio luminoso. Nello stato fondamentale, con la configurazione elettronica del vanadio (III) 3 D 2, sono occupati due elettroni spaiati D Sottolivello ε:

C'è solo due opzioni per la transizione di due elettroni SU D Sottolivello γ: entrambi Entrambi gli elettroni sono occupati Dγ-AO, o solo uno di loro. Qualsiasi altra transizione elettronica associata ad una diminuzione dello spin totale è vietata.
Le transizioni indicate degli elettroni che hanno ricevuto energia in eccesso corrispondono a banda di assorbimento circa 400 nm nello spettro di assorbimento di una soluzione di cloruro di esaacquavanadio(III). L'assorbimento della regione viola-viola dello spettro conferisce un colore aggiuntivo alla soluzione - verde acceso. Se l'agente complessante ha una configurazione elettronica D 0 o D 10 allora transizioni elettroniche Con Dε- acceso D sottolivello γ o viceversa impossibile o perché assenza di elettroni, o perché assenza di orbitali liberi. Pertanto, soluzioni di complessi con agenti complessanti come Sc(III) (3 D 0), Cu(I) (3 D 10), Zn(II) (3 D 10), Cd(II) (4 D 10), ecc., non assorbono energia nella parte visibile dello spettro e appaiono incolore. La selettività dell'assorbimento della luce non dipende solo da agente complessante E il suo stato di ossidazione, ma anche da tipo di ligandi. Quando si sostituiscono i ligandi sul lato sinistro della serie spettrochimica in un composto complesso con ligandi che creano forte campo elettrostatico osservato aumento la frazione di energia assorbita dagli elettroni dalla luce trasmessa e, di conseguenza, diminuire lunghezza d'onda della corrispondente banda di assorbimento. Pertanto, una soluzione acquosa contenente cationi tetraacquarame(II) 2+ è blu e una soluzione di tetraamminorame(II) 2+ solfato è intensamente blu.
Informazioni correlate.
