Begrepet krystallfeltteori. Modeller for kjemisk binding. Krystallfeltteori. Lav- og høyspinnkomplekser
Svakt felt sterkt felt
Midtfelt
Frac34;¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾® Δo
Svake feltligander med elementer fra 3d-serien danner komplekser med høy spinn, og sterkfeltsligander danner komplekser med lavt spinn. Forskjellen mellom dem påvirker den elektroniske strukturen til kompleksene bare for konfigurasjoner d 4 – d 7:
3+ d 5 3– d 5
høyspinn kompleks lavspinn kompleks
H 2 O – svak feltligand CN – – sterk feltligand
Lavspinnkomplekser er alltid mer stabile enn høyspinnkomplekser. Medium-field ligander, avhengig av forhold (ladning og natur av det sentrale atom), kan danne både høy-spinn og lav-spinn komplekser.
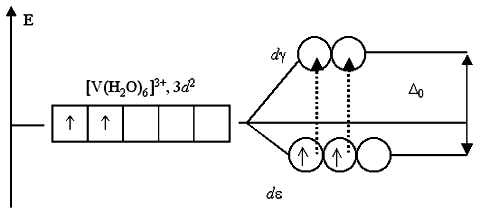
Eksempel. Basert på TCP, foreta en antagelse om den elektroniske strukturen til heksamminkobolt(II) (Δo = 21600 cm–1, P = 21000 cm–1) og heksamminkobolt(III)-ioner (Δo = 9500 cm–1, P = 22500 cm–1).
Ammoniakk er en mellomfeltligand og kan, avhengig av graden av oksidasjon av metallet, danne både høyspinn- og lavspinnkomplekser. La oss finne ut hvilke komplekser som vil være energimessig mer stabile for kobolt(II) og kobolt(III). For å gjøre dette, sammenligne ESC for hvert ion i et sterkt og svakt felt:
(a) 3+, d 6

sterkt felt svakt felt
ESKP (sterkt felt) = –6´(2/5)Δo + 2P = –6´(2/5) ´21600 + 2´21000 = –9840 cm –1
ESKP (svakt felt) = –4´(2/5)Δo + 2´(3/5)Δo = –4´(2/5) ´21600 + 2´(3/5) ´21600 = –8640 cm – 1
Energigevinsten er større ved lavspinnkompleks.
(b) 2+ , d 7

sterkt felt svakt felt
ESKP (sterkt felt) = –6´(2/5)Δo + 1´(3/5)Δo + P = –6´(2/5)´9500 + 1´(3/5) ´9500 + 22500 = 7900 cm–1
ESKP (svakt felt) = –5´(2/5)Δo + 2´(3/5)Δo = –5´(2/5) ´9500 + 2´(3/5) ´9500 = –7600 cm – 1
Energigevinsten er større i tilfellet med et kompleks med høy spinn.
Dermed er 3+-ionet lavspinn og 2+-ionet er høyspinn.
ESC øker med økende Δo, men det er forskjellig for tilstander med høyt spinn og lavt spinn (Fig. 1.28. Avhengigheten av ESC for komplekser med høyt spinn og lavspinn med konfigurasjon d 6 av verdien Δo = 10Dq Regionen der eksistensen av begge stater er mulig er skyggelagt). Regionen nær skjæringspunktet mellom disse to linjene tilsvarer komplekser som kan eksistere i både høyspinn- og lavspinntilstander.
Et eksempel er jern(II)tiocyanatkomplekset med 1,10-fenantrolin, som er høyspinn (paramagnetisk) ved lave temperaturer, og lavspinn (diamagnetisk) ved forhøyede temperaturer (M. Marchivie, P. Guionneau, J.A.K. Howard , G. Chastanet, J.-F. Letard, A. E. Goeta, D. Chasseau, J. Am. Chem. Soc., 2002, v. 124, s. 194). Endringen i multiplisitet er ledsaget av en endring i interatomiske avstander og geometrien til koordinasjonsmiljøet: lavspinnkomplekset er et vanlig oktaeder, og høyspinnkomplekset er forvrengt. Den omvendte overgangen til høyspinntilstanden er mulig under påvirkning av høyt trykk eller stråling. For tiden er flere dusin slike systemer kjent.
Når vi snakket om σ-donor- og π-akseptoregenskapene til liganden, gikk vi utover TCP, ved å bruke tilnærmingene til den molekylære orbitalmetoden som brukes på komplekse forbindelser (volum 1). La oss huske at bildet av spaltningen av d-orbitaler er et fragment av det generelle skjemaet for molekylære orbitaler i et oktaedrisk kompleks, hvor t 2g orbitaler anses som ikke-bindende, og f.eks - som antibinding (fig. bind 1) .
Dannelsen av bindinger i et oktaedrisk kompleks uten π-binding involverer s-, p- og d-orbitalene til metallet og en orbital fra hver ligand. Fra 15 atomorbitaler dannes 15 molekylære orbitaler, seks av dem (a 1 g, t 1 u, e g (fotnote: bokstaven i betegnelsen på orbitaler indikerer graden av deres degenerasjon: t - tre ganger degenerert, e - dobbelt degenerert, a - ikke-degenerert, og tilstedeværelsen av et symmetrisenter: g - symmetrisk, u - asymmetrisk)) σ-binding, tre (t 2 g) - ikke-binding, og seks (f.eks. *, t 1 u *, en 1 g *) σ-løsning. Bindingsorbitaler er nærmere i energi til ligandorbitalene, mens ikke-bindende orbitaler er lokalisert hovedsakelig på metallatomet. Energien d xy , d xz , d yz (t 2 g) til metallorbitalene endres praktisk talt ikke under dannelsen av komplekset.
Tilstedeværelsen av en ledig orbitaler med lav energi i liganden, som i symmetri ligner metallorbitalene, fører til en reduksjon i energien til t 2g orbitaler, praktisk talt uten å påvirke f.eks. og dermed øke Δо (fig. 1.29. Fragmenter av MO diagram for kobolt(III)-komplekset med σ-donorligand (a) og σ-donor, π-akseptorligand (b)).
Jahn-Teller-effekt. I 1937 beviste Yang og Teller teoremet om at ethvert ikke-lineært molekyl i en degenerert elektronisk tilstand er ustabil og spontant gjennomgår en forvrengning som senker symmetrien og fører til fjerning av degenerasjon. Teoremet forutsier bare selve det faktum å fjerne degenerasjon, men indikerer ikke hvordan det vil bli fjernet. Basert på denne teoremet ble forvrengningen av den oktaedriske geometrien til en rekke komplekser forklart, og selve faktumet av tilstedeværelsen av en slik forvrengning ble kalt Jahn-Teller-effekten. La oss se på et eksempel. Kobber(II)-komplekser med d9-konfigurasjon representerer som regel ikke et regulært oktaeder, men er forlenget eller komprimert langs en av aksene (fig. 1.30. Forvrengning av oktaedrisk geometri i kobber(II)-komplekser). La oss vurdere tilfellet med et prolat-oktaeder. Fjerning av ligander plassert langs z-aksen forårsaker fjerning av degenerasjon på grunn av en endring i energiene til orbitalene. Orbitaler rettet langs z-aksen (d xz, d yz, d z 2) samhandler svakere med orbitalene til liganden sammenlignet med orbitaler som ikke har en z-komponent (d xy, d x 2 -y 2), og reduserer derfor energien deres. Et par orbitaler med samme symmetri, som har en z-komponent (d xz, d yz), forblir degenerert og får økt energi. (Fig. 1.31. Endring i energiene til d-orbitaler når oktaederet er forvrengt). Jahn-Teller-effekten manifesterer seg sterkest i komplekser med ulikt fylte e g orbitaler, det vil si med konfigurasjoner t 2g 3 e g 1 (tilsvarende d 4-ionet i et svakt felt: CrCl 2, K 3 MnF 6) og t 2g 6 e g 3 ( tilsvarer d 9-ionet: nesten alle kobber(II)-komplekser) og t 2g 6 e g 1 (tilsvarer d 7-ionet i et sterkt felt, sjelden, K 3 NiF 6),. En ubetydelig Jahn-Teller-effekt er typisk for komplekser med ulikt fylte t 2g orbitaler, det vil si for elektroniske konfigurasjoner t 2g 1 (d 1), t 2g 2 (d 2), t 2g 4 (d 4 i et sterkt felt) , t 2g 5 (d 5 i et sterkt felt), t 2g 5 e g 1 (d 6 i et svakt felt), t 2g 5 e g 2 (d 7 i et svakt felt). Ioner med konfigurasjoner d 3 og d 5 i et svakt felt, d 3 og d 6 i et sterkt felt, d 8 og d 10 er under ingen omstendigheter Jahn-Teller.
Jahn-Teller-effekten manifesterer seg i ulikheten i bindingslengder i mange kobber(II)- og mangan(III)-komplekser og i en ikke-monoton endring i de trinnvise stabilitetskonstantene til kompleksene. For eksempel, i vannfritt kobber(II)klorid er kobberatomet omgitt av seks kloratomer, hvorav fire er lokalisert i en avstand på 0,230 nm, og de to andre er plassert i en avstand på 0,295 nm fra det.
Det er kjent kobber(II)-komplekser (Cl 2, (C 6 H 5 SO 3) 2, etc.), bestående av flere krystallografisk ikke-ekvivalente Jahn-Teller-ioner, hver med sin egen type forvrengning, som omdannes til hverandre, endres metall-ligand-avstanden så fort at totalt sett ser alle metall-ligand-avstander ut til å være de samme. Denne saken ble kalt dynamisk eller pulserende Jahn-Teller-effekt(P.E.M. Wijnands, J.S. Wood, J. Redijk, W.J.A. Maaskant, Inorg. Chem., 1986, 35, 1214).
Jahn-Teller-effekten er imidlertid ikke en universell lov. For tiden er det kjent komplekse ioner med en Jahn-Teller-konfigurasjon, som er uforvrengte oktaedre: 4–, 3+.
Splitting i felt med annen symmetri enn oktaedrisk.
I tillegg til oktaedriske, er det mange kjente komplekser med en annen geometri - kvadratisk plan, tetraedrisk, trigonal-pyramideformet, kvadratisk-pyramideformet, lineær osv. Splittingen i hvert av disse feltene er annerledes enn i oktaederet; det er bestemt av symmetrien til koordinasjonspolyederet.
Square-planare komplekser kan betraktes som et ekstremt tilfelle av tetragonal forvrengning av den oktaedriske geometrien, når ligandene som ligger langs en av koordinataksene fjernes til det uendelige (fig. 1.27b). Betegnelsene til orbitalene er vist i figuren. Planare kvadratiske komplekser er mest typiske for ioner med den elektroniske konfigurasjonen d 8 – Ni 2+, Pd 2+, Pt 2+, Au 3+. Stabiliteten deres øker kraftig med økende Δ, det vil si når de går fra elementer i 3d-serien til tunge overgangselementer. Så, for eksempel, hvis palladium, platina og gull har nesten alle komplekser med et koordinasjonsnummer på fire kvadrater, danner nikkel plane kvadratiske komplekser bare med høyfeltsligander: 2–, Ni(dmg) 2. Nikkel(II)-komplekser med lavfeltligander, slik som halogener, har en tetraedrisk geometri.
Noen kvadrat-planare overgangsmetallkomplekser danner kjeder i fast form med brodannende ligander, for eksempel Pt-CN-Pt i K 2 Br 0,3, hvor platinaatomene er delvis i +4 oksidasjonstilstand. Den høye penetreringsevnen til 5d-orbitaler sikrer deres overlapping med dannelsen av et enkelt energibånd, og følgelig metallisk ledningsevne i kjedens retning. Slike molekylære komplekser er i stand til å lede elektrisk strøm og studeres for tiden intensivt.
I et felt med tetraedrisk symmetri har orbitalene d xy , d yz , d xz maksimal energi, de kalles t 2 -orbitaler, og minimumsenergien er orbitalene d x 2 –y 2 og d z 2, de er betegnet e . På grunn av tilstedeværelsen av et mindre antall ligander og deres forskjellige arrangement, viser det tetraedriske feltet (Fig. 1.32. Sammenligning av spaltninger i de tetraedriske og oktaedriske feltene) seg å være 2,25 ganger svakere enn det oktaedriske: .
De fleste tetraedriske komplekser er høyspinn.(Fotnote - Det er kjent flere eksempler på lavspinn-tetraedriske komplekser, for eksempel Cr(N(Si(CH 3) 3) 2 ) 3 NO (krom(II), d 4 ; D. C. Bradley, Chem. Ber., 1979, 11, 393); CoL 4, hvor L er 1-norbornyl (kobolt(IV), d 5; E.K: Brune, D.S. Richeson, K.H. Theopold, Chem. Commun., 1986 , 1491)). Maksimal stabilisering av det tetraedriske miljøet ved det krystallinske feltet oppnås med konfigurasjoner d 2 (FeO 4 2–, MnO 4 3–) og d 7 (2–). På grunn av den relativt lave stabiliseringsenergien dannes tetraedriske komplekser oftere av ioner med konfigurasjoner d 0 (TiCl 4, MnO 4 –, CrO 4 2–), d 5 i et svakt felt (FeCl 4 –) og d 10 (ZnCl). 4 2–) med null ESKP, samt ikke-overgangsmetallioner (AlCl 4 –). Dannelsen av tetraedriske komplekser sammenlignet med oktaedriske komplekser favoriseres ofte av den steriske faktoren, for eksempel er ionet mer stabilt enn 3–.
Bruke TCP for å forklare stabiliteten til komplekser. Irving-Williams-serien. Krystallfeltteorien gjør det mulig å forklare den ikke-monotoniske naturen til endringer i energiene til krystallgitteret av oksider og halogenider, stabilitetskonstanter for komplekser osv. Rekkefølgen for endring i hydratiseringsenergiene til dobbeltladede kationer av 3d-metaller faller generelt sammen med arten av endringer i ESC i komplekser med høy spinn (fig. 1.33. Endring i hydreringsenergien til dobbeltladede kationmetaller i 3d-serien (a) og endringen i ESC i komplekser med høy spinn (b) ), jo sterkere stabilisering av krystallfeltet, desto større hydrering. Det er kjent at konstantene for substitusjon av et vannmolekyl med en svakfeltligand L
2+ + L x– = (2-x)+ + H 2 O
adlyd Irving-Williams-serien: Mn 2+< Fe 2+ < Co 2+ < Ni 2+ < Cu 2+ < Zn 2+ (Рис. 1.34. Зависимость первой константы устойчивости комплекса от природы 3d-металла). Согласно этому ряду, наибольшей устойчивостью обладают комплексы меди(II) и никеля(II). Простейший вариант ЭСКП предсказывает наибольшую устойчивость никелевых комплексов. При этом надо учитывать, что комплексы меди(II) имеют сильно искаженную октаэдрическую геометрию, что вносит существенный вклад в величину константы устойчивости.
Nepheloauxetisk effekt. Det ble oppdaget at den gjensidige frastøtingen av d-elektroner svekkes når atomet plasseres i liganderfeltet. Denne effekten av liganden på d-elektronene til metallatomet kalles den nefeloauxetiske effekten fra de greske ordene νεφελη - sky og αυξανω - økning. Serien av ligander, arrangert i rekkefølge for å øke deres innflytelse på metallorbitalene, tilsvarer nesten fullstendig den spektrokjemiske serien. Årsaken til den nefeloaksetiske effekten er overlappingen av metallets d-orbitaler med orbitalene til liganden, på grunn av hvilken d-skyen ekspanderer i rommet. Tilstedeværelsen av denne effekten demonstrerer klart begrensningene til den enkleste elektrostatiske modellen - teorien om krystallinsk felt, som antar at lignader er negative punktladninger.
Ligandfeltteori. Krystallfeltteori ble utviklet av Bethe i 1929. For tiden er den mye brukt i modifisert form med korreksjoner for en viss kovalens av metall-ligandbindingen. Denne teorien kalles ligandfeltteori. Tilstedeværelsen av et kovalent bidrag endrer energien til metallorbitalene sammenlignet med den som beregnes av TCP. Andelen av kovalens tas i betraktning ved å innføre korreksjonsfaktorer som gjør det mulig å likestille de eksperimentelle verdiene med de beregnede.
Farging av komplekser.
Fargen på d-overgangselementkomplekser er assosiert med elektronoverganger fra en d-orbital til en annen. Dette er tydelig illustrert av eksemplet med Ti 3+-ionet, omtalt i første bind av læreboken. Ved å absorbere energi som tilsvarer de blå og grønne delene av det synlige spekteret, beveger det eneste d-elektronet i Ti 3+ ion seg til e g orbital (fig. 1.35. Spektrum av 3+ ion). Fargen på ionet skyldes flere farger - rød og fiolett. (Fotnote - Den oppmerksomme leser vil legge merke til en viss asymmetri i absorpsjonsbåndet. Det er en konsekvens av en liten splittelse av t 2g-nivået forårsaket av Jahn-Teller-effekten). Et diagram som viser komplementære farger og som er godt kjent for alle kunstnere, er presentert på andre blad i læreboken. Overgangsenergien, uttrykt i resiproke centimeter (1000 cm –1 = 12 kJ), tilsvarer splittparameteren Δο - den bestemmes oftest fra elektroniske spektre. Bølgelengden er omvendt proporsjonal med energi:
 .
.
Når det gjelder komplekser med et stort antall elektroner, blir spektrumbildet mer komplisert, og ytterligere bånd vises i det. Dette skyldes at den eksiterte tilstanden t 2g 1 e g 1 kan realiseres på flere måter, avhengig av hvilke to d-orbitaler elektronene befinner seg i. For eksempel vil en tilstand der elektroner okkuperer d xy og d x 2 –y 2 orbitaler ha høyere energi enn en d xy 1 d z 2 1 tilstand på grunn av den større frastøtingen av elektroner langs x-aksen. Energien som tilsvarer båndet med lavest energi er lik splitteparameteren Δo.
For å beskrive elektroniske spektre mer detaljert, er det nødvendig å introdusere noen konsepter. La oss kalle ethvert arrangement av elektroner på et undernivå for en mikrotilstand. Antall mikrotilstander N, der n elektroner opptar x orbitaler, er lik

Hver mikrostat er preget av sine egne verdier for spinn og vinkelmomentum. Et sett med mikrotilstander med identiske energier kalles begrep, for eksempel 3 P, 5 D, 1 S. Den digitale indeksen indikerer multiplisitet, som beregnes som:
multiplisitet = antall uparrede elektroner i grunntilstanden + 1.
Navnene på begrepene leses med en indikasjon på multiplisitet: "triplett P", "kvintett D", "singlett S". Bokstaven angir det totale vinkelmomentet L til et atom eller ion, som er lik maksimalverdien av summen av vinkelmomentet m l til individuelle orbitaler okkupert av elektroner. For eksempel inneholder Ti 3+-ionet ett d-elektron, antall mikrotilstander er N = (2´5)!/1!(2´5 – 1)! = 10, L = 2(D) (siden for d-orbitalen m l = –2, –1, 0, 1, 2, er antallet elektroner 1, derfor er den maksimale summen m l lik den største verdien av m l), multiplisitet 1 + 1 = 2. Derfor er grunntilstandsleddet (med lavest energi) 2 D. I tilfellet med et ion med en elektronisk konfigurasjon d 2 N = (2´5)!/2!( 2´5 – 2)! = 45, L = 3(F) (siden for d-orbitalen m l = –2, –1, 0, 1, 2, er antallet elektroner 2, derfor er den maksimale summen av de to største verdiene lik m l), multiplisitet 2 + 1 = 3. Følgelig er termen til grunnmikrotilstanden 3 F. Med et annet arrangement av to elektroner på d-undernivået oppnås tilstander beskrevet av andre termer - 3 P, 1 G , 1 D, 1 S, etc. Forholdet mellom de numeriske verdiene til L og de alfabetiske symbolene er gitt nedenfor:
L = 0 1 2 3 4 5 6 7
På samme måte kan vi utlede vilkårene for grunnen og eksiterte tilstander for andre ioner av d-elementer (tabell 1.5.). Vær oppmerksom på at betingelsene for ioner med konfigurasjon d n og d 10-n er de samme.
Bord. 1.5.
Vilkår for bakken og nærmeste eksiterte tilstander for forskjellige konfigurasjoner av d-elektroner.
Begrepene er delt i det oktaedriske feltet som orbitaler, angitt med lignende bokstaver. D-ledd er delt inn i T 2 g- og E g-komponenter, som d-orbitaler, F-ledd - i T 1 g, T 2 g og A 2 g, som f-orbitaler. S- og P-begrepene er ikke delt i det hele tatt. Mulighetene for elektronoverganger mellom ulike tilstander er begrenset av seleksjonsregler. I komplekser er altså bare overganger mellom tilstander med samme mangfold tillatt. Hver slik overgang tilsvarer et bånd i absorpsjonsspekteret. Som et eksempel kan du vurdere det elektroniske spekteret til kompleks 3+ (fig. 1.36. Elektronisk spektrum av kompleks 3+). De tre båndene skyldes tre elektroniske overganger: 4 A 2 g ® 4 T 2 g, 4 A 2 g ® 4 T 1 g, 4 A 2 g ® 4 T 1 g (P). Overgangen med lavest energi tilsvarer verdien av spalteparameteren: Δo = 17400 cm–1. Komplekset absorberer lys i de røde (17400 cm–1) og blå (23000 cm–1) delene av det synlige spekteret og i nær ultrafiolett (37800 cm–1), derfor har det en fiolett farge.
I følge Laportes regel er overganger mellom stater med samme paritet, som inkluderer s-s, p-p, d-d, f-f overganger, usannsynlige, eller på spektroskopispråket er de forbudt i oktaedriske komplekser. Forbudte overganger er mulige, men skjer med lav intensitet. Dette er grunnen til at overgangsmetallsalter har en merkbar farge bare i konsentrerte løsninger. Det er mange ganger svakere enn fargen på permanganat eller dikromat, hvis ioner ikke inneholder d-elektroner.
Laportes regel gjelder bare for komplekser som har et symmetrisenter. Når oktaederet er forvrengt, forsvinner symmetrisenteret, Laporte-forbudet oppheves, og fargen vises. For eksempel er 3+-ionet fargeløst, men løsninger av jern(III)-salter er ofte gul-oransje på grunn av hydrolyse som fører til dannelse av asymmetriske partikler med et forvrengt oktaedrisk miljø.
Fargen på kompleksene, i tillegg til d-d-overganger fra en metall-d-orbital til en annen (fra t 2g til f.eks. i oktaedriske komplekser), bestemmes av ytterligere to faktorer: overganger fra ligand-orbitaler til metall-orbitaler (de kalles ladningsoverføring). ) og overganger innenfor ligandorbitalene. Disse overgangene faller ikke inn under Laportes styre og har derfor høy intensitet.
Ladningsoverføringsbåndet er tilstede i det elektroniske spekteret til enhver forbindelse, men i noen tilfeller er det i den ultrafiolette delen av spekteret og oppfattes ikke av oss som farge. Hvis forskjellen mellom energiene til ligandorbitalene og metallorbitalene reduseres, faller ladningsoverføringsbåndet inn i den synlige delen av spekteret. Det er ladningsoverføring som forklarer den intense fargen til permanganat, dikromat, kvikksølvsulfid, titan(IV) peroksokomplekser og mange andre forbindelser med tomme d-orbitaler. I noen tilfeller, under påvirkning av lys, skjer ladningsoverføring fra orbitalene til liganden til metallets orbitaler irreversibelt, det vil si at den er ledsaget av en kjemisk prosess. Et eksempel er den fotokjemiske nedbrytningen av sølvhalogenider, som er grunnlaget for svart-hvitt-fotografering: Ag + Br – ¾® Ag 0 + Br 0 .
I det elektroniske spekteret av kaliumpermanganat observeres fire bånd, tilsvarende overganger av elektroner fra ikke-bindende orbitaler lokalisert hovedsakelig på liganden (a 1, t 2 σ orbitaler og e, t 1, t 2 π orbitaler) til e*, t2 '' antibindings orbitaler orbitaler lokalisert på metallatomet ((Fig. 1.37. Energidiagram av det tetraedriske ionet MnO 4 - med π-binding. Elektronoverganger er vist med piler):
ν 1 , Mn(e*) ¾ O(t 1) 17700 cm –1
ν 2 , Mn(t 2 '') ¾ O(t 1) 29500 cm –1
ν 3 , Mn(e*) ¾ O(t 2) 30300 cm –1
ν 4 , Mn(t 2 '') ¾ O(t 2) 44400 cm –1
Båndet med lavest energi faller i den synlige delen av spekteret (λ = 107/17700 = 565 nm), som tilsvarer absorpsjonen av grønt lys og overføringen av karmosinrødt lys.
3. Mekanismer for reaksjoner som involverer komplekse forbindelser.
De aller fleste kjemiske prosesser skjer som en sekvensiell kjede av noen elementære stadier, og reaksjonsligningen inneholder bare informasjon om de viktigste sluttproduktene av reaksjonen. Denne sekvensen av elementære transformasjoner på veien fra utgangsstoffer til produkter kalles en mekanisme. Intermediære, vanligvis ustabile forbindelser som veien fra reaktanter til produkter går gjennom, kalles mellomprodukter. Ethvert mellomprodukt har en viss levetid, vanligvis ekstremt kort, opptil 10 -14 s. På energiprofilen til reaksjonen tilsvarer det et minimum (fig. a) (fig. 1.38. Energiprofiler for en reaksjon som går gjennom: (a) mellomliggende, (b) overgangstilstand.). Som regel kan mellomprodukter påvises i en reaksjonsblanding ved spektrale metoder, og bare i sjeldne tilfeller kan de isoleres i individuell form. Derfor oppnås hovedinformasjonen om reaksjonsmekanismen vanligvis gjennom å studere dens kinetikk - bestemme hastighetskonstanter og beregne aktiveringsparametre (entalpi, entropi, volum). I dette tilfellet er mekanismen en modell som er i samsvar med de kinetiske dataene, en modell som kan forbedres, modifiseres, revideres.
I noen reaksjoner dannes ikke mellomprodukter, og overgangen fra reaktanter til produkter skjer sekvensielt - ett av atomene fjernes gradvis, og de andre nærmer seg. I dette tilfellet sies reaksjonen å fortsette overgangsfase eller aktivert kompleks. Det tilsvarer et maksimum i energiprofilen til reaksjonen (fig. B).
Tillegg: Labile og inerte komplekser
Den termodynamiske stabiliteten til en partikkel bestemmes av endringen i Gibbs-energien for reaksjonen av dens dissosiasjon, eller av verdien av stabilitetskonstanten til denne prosessen. Kinetisk stabilitet viser hvor raskt en gitt partikkel interagerer med andre partikler eller gjennomgår forfall. En kjemisk partikkel vurderes inert, hvis den reagerer med en halveringstid på mer enn 1 minutt. Partikler som reagerer med en høyere hastighet kalles labil. Det må huskes at kinetisk og termodynamisk stabilitet ikke er avhengig av hverandre, det vil si at det samme stoffet kan ha en høy stabilitetskonstant og samtidig være inert, eller omvendt, labilt. Noen slike eksempler er gitt i tabell 1.6.
Tabell 1.6. Stabilitetskonstanter og hastigheter for ligandsubstitusjon i cyanokomplekser av noen metaller.
Henry Taube viste sammenhengen mellom den kinetiske stabiliteten til oktaedriske komplekser og den elektroniske konfigurasjonen av sentralionet i det oktaedriske feltet. I følge Taube er følgende komplekser labile:
· har minst én ledig t 2g orbital - de kan bruke den i reaksjoner i henhold til den assosiative (A, I a) mekanismen, eller
· ha minst ett elektron i e g orbital - dette fremmer reaksjonen ved den dissosiative (D, I d) mekanismen, fordi Å fjerne et elektron fra e g orbital senker energien til overgangstilstanden.
Altså oktaedriske komplekser av krom(III) (t 2g 3), lavspinnkomplekser av jern(II) (t 2g 6) og jern(III) (t 2g 5), samt komplekser av 4d-, 5d- overgangselementer er klassifisert som inerte med antall d-elektroner mer enn to.
SLUTT PÅ TILLEGG
En enhetlig klassifisering av uorganiske reaksjoner er ennå ikke utviklet. Konvensjonelt kan vi foreslå følgende skjema (fig. 1.39. Skjema som illustrerer klassifiseringen av uorganiske reaksjoner):
1) Reaksjoner med substitusjon, addisjon eller eliminering av ligander påvirker en endring i koordinasjonssfæren til metallet,
2) Redoksreaksjoner er assosiert med en endring i den elektroniske konfigurasjonen av metallet, men påvirker ikke koordinasjonsmiljøet,
3) Reaksjoner av koordinerte ligander involverer en endring i liganden uten å endre koordinasjonssfæren til komplekset.
Substitusjonsreaksjoner. I vid forstand betyr substitusjonsreaksjoner prosessene med å erstatte noen ligander i koordinasjonssfæren til et metall med andre. Slike reaksjoner kan skje enten med eller uten endring i oksidasjonstilstanden. Etter klassifiseringen ovenfor vil vi bruke dette begrepet kun i forhold til reaksjoner som skjer uten endring i oksidasjonstilstander.
Klassifiseringen av substitusjonsreaksjoner i uorganisk kjemi ble utviklet av Langford og Gray. Den er basert på definisjonen av den såkalte begrensende mekanismen, og ikke på beskrivelsen av en spesifikk mekanisme. Først bestemmes den støkiometriske mekanismen, og deretter den interne. Støkiometrisk mekanisme er en sekvens av elementære stadier i overgangen fra utgangsstoffer til produkter. Det kan være dissosiativt (D), assosiativt (A) og bytte (gjensidig bytte, I). Dissosiative og assosiative prosesser representerer så å si to begrensende tilfeller, direkte motsatte av hverandre. Begge prosessene skjer i to trinn gjennom dannelsen av et mellomprodukt.
Dissosiativ (D)
Prosessen er to-trinns, i det begrensende tilfellet går den gjennom et mellomprodukt med redusert konsentrasjon:
ML 6 + L, + Y ¾® ML 5 Y
Assosiativ (A)
Prosessen er to-trinns, preget av dannelsen av et mellomprodukt med økt konsentrasjon:
ML 6 + Y, ¾® ML 5 Y + L
Gjensidig utveksling (I)
De fleste utvekslingsreaksjoner foregår gjennom denne mekanismen. Prosessen er ett-trinns og er ikke ledsaget av dannelsen av et mellomprodukt. I overgangstilstanden er reagenset og den avgående gruppen assosiert med reaksjonssenteret, går inn i dens nærmeste koordinasjonssfære, og under reaksjonen blir en gruppe fortrengt av en annen, en utveksling av to ligander skjer:
ML 6 + Y ML 5 Y + L.
Overgangstilstanden er enten et ytre sfærekompleks eller, når det gjelder ladede ligander, et ionepar MX 5 L + Y - .
Intern mekanisme (en eller d) karakteriserer prosessen med ligandsubstitusjon på molekylært nivå. Den viser hvilken av de to prosessene - dannelsen eller bruddet av en binding i overgangstilstanden - som er begrensende. Hvis reaksjonshastigheten bestemmes av dannelsen av en binding mellom reaksjonssenteret og reagenset, snakker vi om assosiativ aktivering. Ellers, når den begrensende faktoren er brudd på forbindelsen mellom reaksjonssenteret og den utgående gruppen, fortsetter prosessen med dissosiativ aktivering. Når vi ser på den støkiometriske mekanismen, er det lett å legge merke til at den dissosiative prosessen alltid tilsvarer dissosiativ aktivering, og den assosiative prosessen alltid tilsvarer assosiativ aktivering, det vil si at konseptet med en intern mekanisme viser seg å være informativt bare i tilfelle av en gjensidig utvekslingsmekanisme - den kan oppstå med både dissosiativ (I d) og assosiativ (I a) aktivering. Når det gjelder den resiproke utvekslingsmekanismen med assosiativ aktivering (Ia), avhenger reaksjonshastigheten av naturen til Y. I overgangstilstanden er metallatomet tett bundet til både den utgående gruppen og den angripende nukleofilen. Et eksempel er prosessen med å erstatte et kloratom med brom og jod i et platinakompleks med dietylentriamin (dien):
Y - ¾¾® + + Cl -
Y = Br, I hastigheter varierer sterkt.
Når det gjelder den resiproke utvekslingsmekanismen med dissosiativ aktivering (I d), er reaksjonshastigheten ikke avhengig av reagenset Y. De angripende og utgående gruppene i overgangstilstanden er svakt bundet til sentralionet. Denne mekanismen brukes til å erstatte vann med amin i vannkomplekser av mange overgangsmetaller, for eksempel nikkel:
2+ + Y ¾¾® 2+ + H 2 O
Y = NH 3 , py-hastighetene er nære.
Studiet av mekanismene for substitusjonsreaksjoner i komplekser av mange metaller er bare i det innledende stadiet. Omfattende informasjon er kun innhentet for kvadratiske plankomplekser av platina og oktaedriske komplekser av krom(III) og kobolt(III). Det kan anses som fast etablert at i platina(II)-komplekser skjer substitusjon i henhold til den assosiative mekanismen (A, Ia) gjennom en mellom- eller overgangstilstand i form av en trigonal bipyramid. Oktaedriske kobolt(III)-komplekser reagerer dissosiativt (D, I d-mekanismer). Spesifikke eksempler på slike reaksjoner vil bli vurdert når kjemien til disse elementene skal beskrives.
Redoksreaksjoner. De fleste redoksprosesser er en kompleks kombinasjon av individuelle elementære stadier, som hver involverer overføring av ett eller, mye sjeldnere, to elektroner. Samtidig overføring av et større antall elektroner i løsninger er umulig.
Enkelt-elektronoverføring kan skje gjennom en av to mekanismer: ytre sfære, det vil si ved tunnelering, eller indre sfære, gjennom en brodannende ligand. Intrasfæremekanismen realiseres i komplekser som inneholder halogenider, hydroksidioner og karboksylgrupper som kan fungere som broer mellom metaller. Et eksempel er reaksjonen mellom pentaminklorkobalt(III) og heksakvakrom(II)-ioner. Prosessen kan grovt deles inn i tre stadier: dannelsen av et heterometallisk kompleks med et brodannende kloridion, elektronoverføring og dekomponering av brokomplekset. Det resulterende 2+-ionet, som er labilt, blir øyeblikkelig til et vannkompleks, og det inerte [(H 2 O) 5 CrCl] 2+ interagerer ikke med vann:

Hvis det ikke er noen partikler i systemet som kan fungere som broer, fortsetter prosessen i den ytre sfæren:
2+ + 3+ = 3+ + 2+ .
Det er spesielt nødvendig å fremheve reaksjonene av oksidativ tilsetning og reduktiv eliminering, diskutert i kapittel 6.
Reaksjoner av koordinerte ligander. Denne gruppen av reaksjoner inkluderer modifikasjonsprosesser av ligander koordinert av et metallion. For eksempel kan diketonatkomplekser, som frie diketoner, nitreres, acyleres eller halogeneres. Det mest interessante og uvanlige eksemplet på reaksjoner av koordinerte ligander er mal syntese– en unik metode for å "sette sammen" en ligand på et metallion. Et eksempel er syntesen av ftalocyaniner fra ftalsyrenitril, som forekommer i nærvær av kobber(II)-ioner, og syntesen av en makrosyklisk Schiff-base fra 2-aminobenzaldehyd, som skjer i nærvær av nikkel(II)-ioner:

I fravær av metall, fortsetter prosessen langs en annen vei, og det ønskede produktet er kun tilstede i en liten mengde i reaksjonsblandingen. Metallionet fungerer i templatsyntese som en matrise ("mal"), stabiliserer et av produktene som er i likevekt med hverandre, og forskyver likevekten mot dannelsen. For eksempel, i reaksjonen X + Y ¾® dannes en blanding av produktene A og B, hvor B, som har lavere energi, dominerer. I nærvær av et metallion dominerer substans A i reaksjonsproduktene i form av et kompleks med M (fig. 1.40. Energidiagram av interaksjonen mellom X og Y i fravær av et metallion (til venstre) og i dets tilstedeværelse (b)).
Spørsmål og oppgaver
1. Hvilken av følgende forbindelser har en perovskittstruktur? BaTiO 3, LiNbO 3, LaCrO 3, FeTiO 3, Na 2 WO 4, CuLa 2 O 4, La 2 MgRuO 6. Tabellen over ioniske radier er gitt i vedlegget. Husk at i komplekse oksidfaser kan B-posisjonene inneholde kationer av to forskjellige metaller.
2. Bruk TCP, avgjør om følgende spineller skal være rette eller inverterte: ZnFe 2 O 4, CoFe 2 O 4, Co 3 O 4, Mn 3 O 4, CuRh 2 O 4.
3. Tiocyanation SCN - har to donorsentre - hardt og mykt. Forutsi hvilken struktur tiocyanatkompleksene av kalsium og kobber(I) vil ha. Hvorfor er det ikke mulig å få tak i kobber(II)tiocyanat?
4. Spekteret til Cr 2+ akva-ionet (grunntilstandsledd 5 D) har to bånd (fig. 1.41. Spektrum av Cr 2+ akva-ionet), selv om det blant leddene til de nærmeste eksiterte tilstandene ikke er ett med samme mangfold. Hva forklarer dette? Hvilken farge har dette ionet?
5. Bruk Δο-verdiene nedenfor, beregne ESC for følgende komplekser i kJ/mol:
(a) 2–, Δο = 15000 cm–1,
(b) 2+, Δο = 13000 cm–1,
(c) 2–, Δο (for 4–)= 21000 cm–1,
Ta paringsenergien lik 19000 cm –1, 1 kJ/mol = 83 cm –1. Beregn deres magnetiske momenter (spinnkomponent).
6. Bruk TCP, forklar hvorfor CN-ionet reagerer med heksakvanikkel(III)-ion for å danne heksacyanoferrat(II), og med heksakvanikkel(II)-ion for å danne tetracyanonickelat(II).
7. Nedenfor er reaksjonskonstantene for sekvensiell erstatning av vann i kobber(II)-akvakomplekset med ammoniakk: K 1 = 2´10 4 , K 2 = 4´10 3 , K 3 = 1´10 3 , K 4 = 2´10 2, K5 = 3´10 –1, K6<< 1. Чем объясняется трудность вхождения пятой и шестой молекул аммиака в координационную сферу меди?
8. Hvordan endres stivheten til kationer når de beveger seg langs en 3d-rad? Stemmer dette med endringsrekkefølgen i stabilitetskonstantene til kompleksene (Irving-Williams-serien, fig. 1.34).
9. Forklar hvorfor det heksakvatiske jern(III)-ionet er fargeløst, og løsninger av jern(III)-salter er farget.
10. Foreslå en mekanisme for reaksjonen 3– + 3– = 4– + 2–, hvis det er kjent at innføring av tiocyanation i løsningen fører til endring i reaksjonshastigheten, og hastigheten er praktisk talt uavhengig av tilstedeværelse av ammoniakk. Gi en forklaring på disse fakta.
Konseptet med endringer i den elektroniske strukturen til overgangsmetallioner under påvirkning av det elektriske feltet av ladede partikler som omgir dem ble foreslått av Becquerel og videreutviklet av H.A. Bethe og J. Van Vleck i begynnelsen XX V. Disse konseptene ble brukt på beskrivelsen av den elektroniske strukturen og egenskapene til komplekse forbindelser bare i midten XX århundre av H. Hartmann og modellen ble kalt "krystallfeltteori" (CFT).
Grunnleggende bestemmelser i TCH for overgangskomplekser d metaller Fig. 24):
1. - Komplekset eksisterer og er stabilt på grunn av den elektrostatiske interaksjonen mellom kompleksdanneren og liganden.
2. - Ligander betraktes uten å ta hensyn til deres elektroniske struktur som punktladninger eller dipoler.
3. - Under påvirkning av det elektriske feltet til liganden, degenererer valens fem ganger ( n -1) d orbitaler deles avhengig av symmetrien til ligandmiljøet.
4. - Fordeling av metallvalenselektroner mellom splittede ( n -1) d orbitaler avhenger av forholdet mellom spinn-paringsenergien og splittende energi.
Tenk for eksempel på endringen i energien til femdobbelt degenerert ( n -1) d orbitaler til det sentrale metallionet M n+ , plassert i sentrum av koordinatene, under påvirkning av det oktaedriske feltet av negativt ladede ligander [ ML 6] z , plassert på koordinataksene (fig. 25). Som et resultat av frastøtingen av valenselektronene til metallet fra negativt ladede ligander med en jevn fordeling av negativ ladning rundt metallet (sfærisk symmetrisk elektrisk felt), energien til alle fem d orbitaler vil øke med mengden E 0 sammenlignet med fri M n+ ion. Fordi det d orbitaler har forskjellige romlige orienteringer, så med konsentrasjonen av negative ladninger på ligander plassert på koordinataksene, er økningen i deres energi forskjellig. Energi Boost d z 2 og d x 2- y 2 orbitaler rettet mot liganden på koordinataksene har en større energiøkning dxy, dxz og dyz orbitaler rettet mellom koordinatakser.
Energi av fisjonfemdobbelt degenerert ( n -1) orbitaler til dobbelt degenerert d x 2- y 2, z 2 orbitaler og trippel degenerert d xy, xz, yz orbitaler kalles (fig. 26) krystallfeltsdelingsparameter. Siden energien til splittelsen d orbitaler i det oktaedriske feltet til liganden endres ikke sammenlignet med det sfærisk symmetriske elektriske feltet, så økningen i energien til de to d x 2- y 2, z 2 orbitaler oppstår ved 0,6D 0 og en reduksjon i energien på tre d xy , xz , yz orbitaler med 0,4 D 0 .
For å indikere graden av degenerasjon og symmetri av metallorbitaler delt under påvirkning av det elektriske feltet til ligander, brukes spesielle symboler. Trippel degenerert og symmetrisk med hensyn til symmetrisenter og rotasjon rundt koordinataksene d xy, xz, yz t 2 g ", mens den er dobbelt degenerert og også symmetrisk med hensyn til symmetrisenteret d x 2- y 2, z 2 orbitaler er betegnet med symbolet " f.eks " Således, under påvirkning av det oktaedriske elektriske feltet til liganden, femdoblet degenerert ( n -1) d orbitalene til det kompleksdannende middelet er delt opp i tredobbelt og dobbelt degenererte orbitaler med forskjellige energier t 2 g og e g orbitaler.
En lignende vurdering av endringen i energi av femdobbelt degenerert ( n -1) d orbitaler av et fritt metallion i et tetraedrisk miljø av ligander i [ ML4]z komplekser viser (fig. 27) deres splittelse også i todelt (e) og tredelt ( t ) degenererte orbitaler, men med motsatt energiposisjon. Subscript " g " når betegnet "e" og " t » orbitaler er ikke indikert siden det tetraedriske komplekset ikke har et symmetrisenter. En reduksjon i antall ligander i et tetraedrisk kompleks sammenlignet med et oktaedrisk kompleks fører til en naturlig reduksjon in:D T = 4/9 D OM .
Reduserer symmetrien til ligandmiljøet til metallet, for eksempel tetragonal forvrengning av oktaedrisk [ ML 6] z komplekser assosiert med forlengelse av metall-ligandbindinger med aksiale ligander [ ML 4 X 2] z og dannelsen i det begrensende tilfellet med plan-kvadrat [ ML4]z komplekser, fører (fig. 28) til ytterligere spaltning av valens ( n -1) d metall orbitaler.
Fylling av splitt ( n -1) d metallorbitaler oppstår i samsvar med Pauli-prinsippene og minimumsenergi. For oktaedriske komplekser med d 1 , d 2 og d 3 elektronisk konfigurasjon av metallet, valenselektroner, i samsvar med Hunds regel, befolker t 2 g orbitaler med parallelle spinn, som fører til t 2 g 1 , t 2 g 2 og t 2 g 3 elektronisk struktur av komplekser.
For metaller med d 4 elektronisk konfigurasjon, tre elektroner fylles også t 2 g orbitaler med parallelle spinn. Populasjonen av det fjerde elektronet avhenger av energikostnadene for verdien av spinnparingsenergien (E sp.-sp.) under populasjonen t 2 g orbitaler med antiparallell spinn og brudd på Hunds regel, eller overvinner energien til splitting av krystallfeltetD o ved innsjekking f.eks orbitaler med parallelt spinn i henhold til Hunds regel. I det første tilfellet dannes et kompleks med t 2 g 4 elektronisk struktur og redusert spinn multiplisitet sammenlignet med fritt metall 2 S +1 = 3 (S - totalt spinn), kalt lavt spinn. Når Hunds regel er oppfylt og det fjerde elektronet er befolket på f.eks orbitaler dannes et kompleks med t 2 g 3 e g 1 elektronisk struktur og gratis metalllignende spinn multiplett 2 S +1 = 5. Slike komplekser kalles høyspinn.
Tilsvarende ved fordeling av valens d5, d6 og d7 metallelektroner t 2 g og f.eks orbitaler av oktadriske komplekser avhengig av forholdet E sp.-sp. OgD O Dannelsen av to typer komplekser er mulig:
Ved E sp.-sp. > D O høyspinnkomplekser med metallets elektroniske struktur dannes t 2 g 3 e g 2 , t 2 g 4 e g 2 , t 2 g 5 e g 2 i henhold til Hunds regel og gratis metalllignende spinn multiplisitet - 2 S+1 = 6, 5, 4;
E sp.-sp.< D O lavspinnkomplekser med metallets elektroniske struktur dannes t 2 g 5 e g 0 , t 2 g 6 e g 0 , t 2 g 6 e g 1 og lavere spinn multiplisitet sammenlignet med gratis metall 2 S +1 = 2, 1, 2.
Metallkomplekser med d 8, d 9 og d 10 elektronisk konfigurasjon er preget av én type elektronfordeling - t 2 g 6 e g 2 , t 2 g 6 e g 3 , t 2 g 6 e g 4 med spinn multiplisitet som ligner på gratis metall: 2 S +1 = 3, 2 og 0.
Så parameterenD, som karakteriserer delingen ( n -1) d metallorbitaler under påvirkning av det elektriske feltet til liganden er en av hovedkarakteristikkene til endringer i egenskapene til komplekser sammenlignet med et fritt metallion. Det er parameterverdienDbestemmer for en rekke elektroniske konfigurasjoner av metallet bestemmer muligheten for dannelse av høy- eller lavspinnkomplekser med forskjellige fordelinger av elektroner over delte orbitaler og forskjellige egenskaper.
Verdien tilDavhenger av arten av metallet til kompleksdannende middel, liganden som omgir det og deres romlige posisjon rundt kompleksdannende middel:
1. Ligander i rekkefølge med økende parameterDfor komplekser av samme metall og lignende geometrisk struktur er plassert i den såkalte spektrokjemiske serien: JEG -< Br - < Cl - < F - < OH - < C 2 O 4 2- ~ H 2 O < NCS - < NH 3 ~ En < NO 2 - < CN - < CO . I begynnelsen av raden er det "svake felt"-ligander - halogenidioner, hydroksyd- og oksalationer, vann, som hovedsakelig danner komplekser med høy spinn. Ligandene på høyre side av serien: karbonmonoksid, cyanid og nitrittioner kalles "høyfelt"-ligander og er typisk preget av dannelsen av lavspinnkomplekser. For ligander i midten av serien - tiocyanation, ammoniakk, etylendiamin, avhengig av metallets natur, dannes komplekser med høy eller lav spinn.
2. Øke effektiviteten av det elektriske feltet til ligander på d metallorbitaler med økende størrelse i rad 3 d<< 4 d < 5 d , så vel som en økning i graden av oksidasjon av metallet fører til en økning i parameterenD i serien: Mn(II)< Ni (II ) < Co (II ) < Fe (II ) < V (II ) < Fe (III ) < Co (III ) < Mn (IV ) < Mo (III ) < Rh (III ) < Ru (III ) < Pd (IV ) < Ir (III ) < Pt (IV ).
3. Parameter Dfor tetraedriske komplekser er bare 4/9 av parameterenDoktaedriske komplekser.
"Tunge" komplekser 4 d og 5 d metaller, nesten uavhengig av ligandens natur, danner hovedsakelig lavspinnkomplekser, mens dannelsen av lav- eller høyspinnkomplekser er "lette" 3 d metaller bestemmes hovedsakelig av styrken til ligandfeltet.
I motsetning til MMS, rettferdiggjør krystallfeltteorien forskjellen i de magnetiske egenskapene til komplekser av samme metallion med forskjellige ligandmiljøer, for eksempel diamagnetiske [ Fe(CN ) 6 ] 4- og paramagnetisk [ Fe(H2O ) 6 ] 2+ bruker ikke hypotesen om deres intraorbitale ( d 2 sp 3 hybridisering) og ytre orbital ( sp 3 d 2 hybridisering) struktur. Forskjellen i magnetiske egenskaper bestemmes av lav- og høyspinn-naturen til fordelingen av 6-valente elektroner Fe(II ) ved splitt t 2 g og f.eks orbitaler (fig. 29). Å være sterke og svake feltligander dannes cyanidioner og vannmolekyler Fe(II ) lav- og høyspinnkomplekser med t 2 g 6 e g 0 og t 2 g 4 e g 2 distribusjon av elektroner, som bestemmer diamagnetisme [ Fe(CN ) 6 ] 4- og paramagnetisme [ Fe(H2O ) 6 ] 2+ komplekser.
Splitting av femdobbelt degenerert ( n -1) d metallorbitaler i komplekser og parameterendringerDavhengig av arten av liganden, bestemmer den den karakteristiske fargen på kompleksene både i fast tilstand og i løsninger. Når komplekset absorberer elektromagnetisk stråling i det synlige området av spekteret (400-750) nm, hvor energien til kvanten er E lik verdien D, skjer elektronoverføring fra t 2 g på f.eks orbitaler. Det er den uabsorberte elektromagnetiske strålingen i det synlige området av spekteret som bestemmer fargen på komplekset i samsvar med "Newtons fargesirkel" (fig. 30), som viser primær- og sekundærfargene til synlig stråling.
Aquacomplex titan ( III) [Ti (H 2 O) 6] 3+ c t 2 g 1 e g 0 elektronisk distribusjon som et resultat av fotoeksitasjon, tilsvarende overgangen av elektronet til høyere energi f.eks. orbitaler:
3+ (t 2g 1 e g 0) + hn= * 3+ (t 2g 0 e g 1)
absorberer lyskvanter i det gule området av spekteret, noe som fører til dens fiolette farge. En endring i ligandmiljøet til metallionet i samsvar med posisjonen til liganden i den spektrokjemiske serien fører til en endring i parameterenDog, som en konsekvens av dette, til en endring i energien og bølgelengden til kvanter absorbert av komplekset og til den karakteristiske fargen til komplekset - for eksempel i serien [ CuCl 4 ] 2- , [ Cu (H 2 O ) 4 ] 2+ , [ Cu (NH 3 ) 4 ] 2+ fargen på kompleksene endres fra grønt til blått og fiolett.
Sammen med krystallfeltet splittende energiD, spiller også en viktig rolle i TCH krystallfeltstabiliseringsenergi(ESKP) -økning i energi når du fordeler elektroner blant de som er delt i komplekset ( n -1) d metallorbitaler sammenlignet med energien til fem ganger degenerert ( n -1) d metallorbitaler i et ekvivalent sfærisk elektrisk felt (fig. 31, 32).
ESCP av oktadrale og tetraedriske komplekser.|
Mn+ |
Oktaedriske komplekser |
Tetraedriske komplekser |
|
|
Lavt spinn |
Høy spinn |
Høy spinn |
|
|
0.4 D o |
0.6 D T |
||
|
0.8 D o |
1.2 D T |
||
|
1.2 D o |
0.8 D T |
||
|
d 4 |
1.6 D o |
0.6 D o |
0.4 D T |
|
d 5 |
2.0 D o |
0 D o |
0 D T |
|
d 6 |
2.4 D o |
0.4 D o |
0.6 D T |
|
d 7 |
1.8 D o |
0.8 D o |
1.2 D T |
|
d 8 |
1.2 D o |
0.8 D T |
|
|
d 9 |
0.6 D o |
0.4 D T |
|
|
d 10 |
0 D o |
||
Et estimat av EXP-verdien til komplekset oppnås på grunnlag av splittediagrammer ( n -1) d metallorbitaler i det elektriske feltet til ligander, som viser en reduksjon eller økning i energien til systemet sammenlignet med et sfærisk elektrisk felt når elektroner befolker seg splittet ( n -1) d orbitaler. For oktaedrisk [ ML 6] z komplekser (fig. 32) populasjon av hvert elektron t 2 g orbitaler fører til en økning i systemenergi med 0,4Då, sjekke inn f.eks krever energiforbruk 0,6D O . For tetraedrisk [ ML4]z komplekser med motsatte energiposisjoner e og t metallorbitaler - okkupasjon av hvert elektron ved spaltning e og t orbitaler er ledsaget av en reduksjon og økning i energien til systemet med 0,6D t og 0,4 D T .
Som en refleksjon av den termodynamiske stabiliteten til kompleksene, er estimater av deres ESCR-verdier i samsvar med eksperimentelle data om endringer i energien til krystallgitteret for høyspinn heksafluoridkomplekser 3 d metaller (fig. 33).
ESC-verdier lar oss bestemme den mest foretrukne koordinasjonsisomeren (fig. 34), for eksempel [ Cu (NH 3 ) 6 ][ NiCl 4 ] eller [ Ni (NH 3 ) 6 ][ CuCl 4 ]. For å gjøre dette, beregne forskjellen i ESC for det komplekse kationet og anionet til isomerene. ESCR-verdi [ Cu (NH 3 ) 6 ] 2+ og [NiCl 4 ] 2- er 0,6 D o og 0,8 D T hhv. VurdererD t = 4/9 D o , forskjellen mellom ESCP-verdiene [ Cu (NH 3 ) 6 ] 2+ og [NiCl 4 ] 2- blir 19/45D o . På samme måte er verdiene til ESKP [ Ni (NH 3 ) 6 ] 2+ og [CuCl 4 ] 2- er 1,2 D o og 0,4 D T , og forskjellen mellom dem er 28/45D o . Stor forskjell ESCP kompleks kation [ Ni (NH 3 ) 6 ] 2+ og anion [CuCl 4 ] 2- sammenlignet med [ Cu (NH 3 ) 6 ] 2+ og [NiCl 4 ] 2- viser en mer foretrukket dannelse av isomeren med sammensetning [ Ni (NH3)6][CuCl4].
Sammen med de magnetiske og optiske egenskapene til påvirkningen av den elektroniske strukturen til metallet på den termodynamiske stabiliteten til kompleksene, forutsier TKP en forvrengning av den geometriske strukturen til kompleksene med en ujevn fordeling av elektroner over delt ( n -1) d metallorbitaler (fig. 35). I motsetning til den vanlige oktaedriske strukturen [ Co (CN) 6 ] 3- s t 2 g 6 e g 0 elektronisk distribusjon, tetragonal forvrengning av et lignende kompleks [ Cu (CN) 6 ] 4- s t 2 g 6 e g 3 elektronisk distribusjon som inneholder 3 elektroner på 2 ganger degenerert f.eks orbitaler, fører til effektiv transformasjon av oktaedralen til et kvadratisk-plankompleks:
4- = 2- + 2CN - .
Alt det ovennevnte viser at den relative enkelheten og de brede egenskapene til TCT for å forklare og forutsi de fysisk-kjemiske egenskapene til komplekser bestemmer den store populariteten til denne modellen for å beskrive kjemiske bindinger i komplekse forbindelser. Samtidig, med fokus på endringer i den elektroniske strukturen til metallet under kompleksdannelse, tar TCP ikke hensyn til den elektroniske strukturen til liganden, og vurderer dem som punkt negative ladninger eller dipoler. Dette fører til en rekke begrensninger ved TCP når man skal beskrive den elektroniske strukturen til komplekser. For eksempel, innenfor rammen av TCP er det vanskelig å forklare posisjonen til en rekke ligander og metaller i spektrokjemiske serier, som er assosiert med en viss grad av kovalens og muligheten for dannelse av flere metall-ligandbindinger. Disse begrensningene elimineres når man vurderer den elektroniske strukturen til komplekse forbindelser ved å bruke den mer komplekse og mindre visuelle metoden for molekylære orbitaler.
Valensbindingsteori var den første av de kvantemekaniske teoriene som ble brukt til å omtrent forklare naturen til kjemiske bindinger i komplekse forbindelser. Søknaden var basert på ideen om giver-akseptor mekanisme dannelse av kovalente bindinger mellom liganden og kompleksdanneren. Ligand teller donorpartikkel, i stand til å overføre et par elektroner akseptor – kompleksdannende middel, som gir frie kvanteceller (atomorbitaler) av sine energinivåer for dannelse av bindinger.
For dannelse av kovalente bindinger mellom kompleksdannende middel og ligander, er det nødvendig at den ledige s-, s- eller d-atomiske orbitaler av kompleksdannende middel har gjennomgått hybridisering en bestemt type. Hybride orbitaler opptar en viss posisjon i rommet, og antallet deres tilsvarer koordinasjonsnummer kompleksdannende middel.
Dette skjer ofte kombinere uparede elektroner kompleksdannende middel i par, som tillater frigjøring av et visst antall kvanteceller - atomorbitaler, som deretter deltar i hybridisering og dannelse av kjemiske bindinger.
De ensomme elektronparene til liganden samhandler med hybridorbitalene til det kompleksdannende middelet, og overlapp korresponderende orbitaler av kompleksdannende middel og ligand med tilsynekomsten av økt elektrontetthet i det indre nukleære rommet. Elektronparene til det kompleksdannende middelet samhandler på sin side med de ledige atomorbitalene til liganden, å styrke forbindelsen gjennom dativmekanismen. Dermed er kjemisk binding i komplekse forbindelser vanlig kovalent tilstrekkelig tilkobling varig Og energisk gunstig.
Elektronpar lokalisert i hybridorbitalene til kompleksdannende middel har en tendens til å innta en posisjon i rommet der deres gjensidige frastøting er minimal. Dette leder til struktur komplekse ioner og molekyler ser ut til å være i en viss avhengighet av type hybridisering.
La oss vurdere dannelsen av noen komplekser fra synspunktet til teorien om valensbindinger. Først og fremst bemerker vi at valensorbitalene til atomene til de kompleksdannende midlene er nære i energi:
E (n- 1)d » E ns » E n.p. » E nd
|
Hybridiseringstype |
Geometrien til komplekset |
||
|
lineær |
-
|
||
|
trekantet |
- |
||
|
tetraeder |
2-
|
||
|
2-
|
|||
|
sp 3 d(z 2) |
trigonal bipyramide |
||
|
sp 3 d(x 2 - y 2) |
firkantet pyramide |
3-
|
|
|
sp 3 d 2 , |
3+ |
||
|
sp 3 d 3 |
femkantet bipyramide |
4-
|
For eksempel inkluderer 2+-kationen det kompleksdannende middelet sink(II). Elektronskallet til dette konvensjonelle ionet har formelen 3 d 10 4s 0 4s 0 og kan konvensjonelt avbildes som følger:
Ledig 4 s- og 4 s-orbitaler av sink(II)-atomet danner fire sp 3-hybride orbitaler orientert mot toppene til tetraederet.
Hvert ammoniakkmolekyl har et enkelt elektronpar på nitrogenatomet. Orbitalene til nitrogenatomer som inneholder ensomme elektronpar overlapper med sp 3-hybride orbitaler av sink(II), som danner et tetraedrisk komplekskation av tetraedramin sink(II)2+: 
Siden 2+-ionet ikke har noen uparrede elektroner, viser det seg diamagnetisk egenskaper.
Tetrakloromanganat(II)ion 2- inneholder fem uparrede elektroner per 3 d-orbitaler og ledige 4 s- og 4 s-orbitaler. Det dannes ledige orbitaler sp 3-hybride orbitaler som overlapper med s-atomorbitaler av kloridioner: 
Det tetraedriske ion 2- således oppnådd er paramagnetisk, siden den inneholder fem uparrede elektroner.
Bruke en konvensjonell prediksjonsalgoritme type hybridisering av atomorbitaler innenfor rammen av valensbindingsmetoden kan man bestemme geometri av komplekser av ulik sammensetning. For å gjøre dette er det først og fremst nødvendig å skrive den elektroniske formelen for valensnivået og konstruere et diagram over fordelingen av elektroner over kvanteceller. For eksempel, for et nøytralt nikkelatom:

Overgang 4 s-elektroner for 3 d-undernivå transformerer paramagnetisk Ni-atom 0 tommer diamagnetisk partikkel Ni*: 
De resulterende ledige orbitalene gjennomgår hybridisering, og danner en tetraedrisk konfigurasjon. Bygget slik tetraedrisk diamagnetisk tetrakarbonylnikkelkompleks (CN = 4), som er preget av betydelig stabilitet.
Hvis kompleksdanneren er nikkel(II) med elektronkonfigurasjon 3 d 8 4s 0 4s 0, så behovet for å flytte elektroner fra 4 s-subnivå før hybridisering forsvinner, siden det er et tilstrekkelig antall ledige orbitaler til å realisere koordinasjonsnummer 4: 
Denne strukturen har en ustabil paramagnetisk tetrabromikkolat(II)-ion 2-kompleks. Men når du kombinerer to elektroner 3 d-subnivå til et par og transformasjonen av en av kvantecellene i dette undernivået til en ledig en endrer både typen hybridisering og egenskapene til det resulterende komplekset: 
Hybridiseringstype dsp 2 og den plane kvadratiske formen til komplekset realiseres ved dannelsen av en stall diamagnetisk kompleks tetracyanoniccolate(II)-ion 2- (CN = 4): 
Hvis syntesen av cyanidkomplekset utføres under forhold med overskudd av ligand, kan et koordinasjonsnummer på 5 realiseres: 
Stabil diamagnetisk pentacyanoniccolate(II)-ion 3- komplekset har form av en firkantet pyramide: 
Oktaedrisk nikkel(II) 2+ kompleks, men paramagnetisk, men ganske stabil. Utdanningen hans er på grunn sp 3 d 2-hybridisering av nikkel-atomorbitaler: 
Hvis atomorbitaler av den ytre d-undernivå, kompleks, som regel, i stor grad paramagnetisk og kalles ytre orbital eller høyspinn. Strukturen til slike komplekser kan tilsvare typen hybridisering, for eksempel, sp 3 d 2 .
Slike komplekser, under dannelsen av hvilke hybridisering finner sted med deltakelse av atomorbitaler til de ytre d-undernivåer kalles intra-orbital eller lavt spinn og som regel diamagnetisk eller svakt paramagnetisk(alle eller nesten alle elektronene i kompleksdannende middel er sammenkoblet, og typen hybridisering, f.eks. d 2 sp 3 eller dsp 2).
Ved undersøkelse av jern(II)-komplekser finner man både ytre orbitale og intraorbitale komplekser.


Diagrammet nedenfor viser hvordan de er dannet paramagnetisk høyspinn heksafluorferrat(II)-ion 4- og diamagnetisk lavspinn heksacyanoferrat(II)ion 4-.
Teorien om valensbindinger i seg selv svarer ikke på spørsmålet om hvilken type kompleks som dannes i hvert enkelt tilfelle, siden denne metoden ikke tar hensyn til påvirkningen av ligandens natur. Derfor må valensbindingsmetoden nødvendigvis suppleres med data om kompleksets magnetiske egenskaper eller informasjon om ligandens påvirkning på det dannede kompleksets natur.
.Krystallfeltteori erstattet teorien om valensbindinger på 40-tallet av XX-tallet. I sin rene form brukes den ikke for tiden, siden den ikke kan forklare dannelsen av kovalente bindinger i komplekse forbindelser og ikke tar hensyn til den sanne tilstanden til liganden (for eksempel deres faktiske størrelser) selv i tilfelle av nære interaksjoner til rent elektrostatisk.
Allerede på midten av 50-tallet ble den forenklede krystallfeltteorien erstattet av en forbedret ligandfeltteori, tar hensyn til den kovalente naturen til de kjemiske bindingene mellom kompleksdannende middel og liganden.
Imidlertid er den mest generelle tilnærmingen til å forklare dannelsen av komplekse forbindelser gitt av molekylær orbital teori(MO), som for tiden råder over alle andre. Den molekylære orbitalmetoden sørger for både ren elektrostatisk interaksjon i fravær av overlappende atomorbitaler, og hele settet med mellomliggende grader av overlapping.
La oss se på de grunnleggende konseptene krystallfeltteori, som i likhet med teorien om valensbindinger fortsatt beholder sin betydning for den kvalitative beskrivelsen av kjemiske bindinger i komplekse forbindelser på grunn av sin store enkelhet og klarhet.
I krystallfeltteori vurderes den kjemiske bindingen mellom kompleksdannende middel og liganden elektrostatisk. I følge denne teorien er ligandene lokalisert rundt det kompleksdannende middelet ved toppunktene til vanlige polyedre ( polyedre) som punktgebyrer. Teorien tar ikke hensyn til det faktiske volumet av liganden.
Ligander, som punktladninger, skaper rundt kompleksdannende middel elektrostatisk felt("krystallfelt", hvis vi vurderer en krystall av en kompleks forbindelse, eller ligandfelt), der energinivåene til kompleksdanneren og fremfor alt, d-undernivåer deler seg, og energien deres endres. Arten av splitting, energien til nye energinivåer avhenger av symmetri arrangement av ligander (oktaedrisk, tetraedrisk eller annet krystallfelt). Når molekylene H 2 O, NH 3 , CO og andre er koordinert som ligander, anses de som dipoler, orientert med en negativ ladning mot kompleksdanneren.
La oss vurdere tilfellet med et oktaedrisk arrangement av ligander (for eksempel 3- eller 3+). I midten av oktaederet er det et kompleksdannende atom M(+n) med elektroner på d-atomiske orbitaler, og på toppene er det ligander i form av punkt negative ladninger (for eksempel F - ioner eller polare molekyler som NH 3). I et konvensjonelt ion M(+n) som ikke er assosiert med ligander, er energiene til alle fem d-AO er de samme (dvs. atomorbitaler degenerert).
Imidlertid i det oktaedriske feltet av ligander d-AOs av kompleksdanneren faller inn i ulik posisjon. Atomiske orbitaler d(z 2) og d(x 2 -
y 2), langstrakt langs koordinataksene, kommer nærmest ligandene. Mellom disse orbitalene og ligandene lokalisert ved toppunktene til oktaederet, oppstår det betydelige forskjeller frastøtende krefter, som fører til en økning i orbital energi. Disse atomorbitalene er med andre ord underlagt maksimal eksponering for ligandfeltet. En sterkt komprimert fjær kan tjene som en fysisk modell for slik interaksjon. 

Tre andre d-AO - d(xy), d(xz) Og d(yz), plassert mellom koordinataksene og mellom liganden, er i større avstand fra dem. Samspillet mellom slike d-AO med ligander er minimal, og derfor energi d(xy), d(xz) Og d(yz)-AO reduseres sammenlignet med den opprinnelige. 

Dermed femdoblet degenerert d-AO kompleksdannende middel, kommer inn oktaedrisk ligandfelt, utsatt splitting i to grupper av nye orbitaler – tre ganger degenererte orbitaler med lavere energi, d(xy), d(xz) Og d(yz), Og dobbelt degenererte orbitaler med høyere energi d(z 2) og d(x 2 -
y 2). Disse nye gruppene d-orbitaler med Nedre Og høyere energi betegne d e og d g: 
Energiforskjell to nye undernivåer d e og d g fikk navnet deleparameter D0:
E 2 – E 1 = D0
Plassering av to nye energi subnivåer d e og d g i forhold til originalen ( d-AO) på energidiagrammet asymmetrisk:
(E 2 – E 0) > (E 0 – E 1).
Kvantemekanisk teori krever at når nye energinivåer er fullstendig befolket med elektroner, forblir den totale energien uendret, dvs. hun burde bli lik E 0 .
Likheten skal med andre ord tilfredsstilles
4(E 2 – E 0) = 6(E 0 – E 1),
hvor 4 og 6 – maksimum antall elektroner pr d g - og d e -AO. Av denne likestillingen følger det at
(E 2 – E 0) / (E 0 – E 1) = 3/2 og
(E 2 – E 1) / (E 0 – E 1 >) = 5/2, eller
D0/( E 0 – E 1) = 5/2, hvorfra ( E 0 – E 1) = 2/5 ´ D 0 >. Plasser hvert elektron ut av de maksimale seks mulige på d e-orbitaler årsaker avta (gevinster) energi med 2/5 D 0 . Tvert imot, plasseringen av hvert elektron av fire mulig på d g orbitaler årsak øke (koste) energi med 3/5 D 0 . Hvis befolket med elektroner d e - og d g -orbitaler helt, så nei vinne energi vil ikke(akkurat som det ikke vil ekstra energiforbruk): 4 ´ 3/5 ´ D 0 - 6 ´ 2/5 ´ D 0 = 0. Men hvis originalen d-AO er kun befolket delvis og inneholder fra 1 til 6 elektroner, og disse elektronene plasseres kun på d e -AO, så får vi betydelig energigevinst. Spesifisiteten til hver ligand påvirker feltet som denne liganden skaper - sterk eller svak. Hvordan sterkere felt ligander enn mer betydning deleparameter D0. Studiet av splittingsparameteren er vanligvis basert på spektroskopisk forskning. Bølgelengder absorpsjonsbånd komplekser l i krystallinsk tilstand eller i løsning, på grunn av overgangen av elektroner fra d e - på d g-AO, assosiert med deleparameter D 0 som følger: n = 1/l; D hvor er Plancks konstant h lik 6.626 ´ 10 - 34 J. s; Delingsparameter, i tillegg til typen ligand, avhenger på graden av oksidasjon Og natur kompleksdannende middel. På økende atomladning av det kompleksdannende atomet D 0 øker også. Hexaamminiridium(III) 3+, hexaamminiridium(III) 3+ og hexaamminiridium(III) 3+ kationer ( Z= 27, 45 og 77) er karakterisert ved å dele parametere lik 22900, 34100 og 41000 cm -1. Avhengigheten av D0 av arten av liganden er mer mangfoldig. Som et resultat av studiet av en rekke komplekse forbindelser, ble det funnet at når det gjelder deres evne til å øke spaltningsparameteren til kompleksdannende metaller lokalisert i deres vanlige oksidasjonstilstander, kan de vanligste ligander ordnes i følgende spektrokjemisk serie, langs hvilken verdien av D 0 øker monotont: Dermed det sterkeste elektrostatiske feltet rundt kompleksdanneren og den sterkeste spaltningen d-AO er forårsaket av NO 2 - ligander, CN -
og CO. La oss vurdere fordelingen av elektroner over d e - og d g-orbitaler i det oktaedriske feltet av ligander. Innsjekking d e - og d g-orbitaler oppstår i full overensstemmelse med Hunds regel Og Pauli-prinsippet. I dette tilfellet, uavhengig av verdien av splittingsparameteren, er de tre første elektronene okkupert av kvanteceller d e-undernivå: Dersom antall elektroner pr d- det er mer enn tre undernivåer av kompleksdanneren; det er to muligheter for å plassere dem på delte undernivåer. Ved en lav verdi av spaltningsparameteren (svake felt av liganden), overvinner elektroner energibarrieren. d e - og d g-orbitaler; det fjerde og deretter det femte elektronet befolker kvanteceller d g-undernivå. Med et sterkt ligandfelt og høy D0-verdi er populasjonen befolket av det fjerde og femte elektronet d g-undernivå ekskludert; utfylling pågår d e-orbitaler. På svake feltligander fylle kvanteceller med 4 eller 5 elektroner parallelle spinn, så det resulterende komplekset viser seg å være sterkt paramagnetisk. I et sterkt ligandfelt ett og deretter to elektronpar dannes på d e -undernivå, altså paramagnetisme komplekset viser seg å være mye svakere. Det sjette, syvende og åttende elektronet i tilfelle av et svakt felt ender opp igjen d e -undernivå, som komplementerer konfigurasjonene til elektronpar (ett i tilfellet d 6, to – d 7 og tre - d 8): Ved et sterkt ligandfelt fyller det sjette elektronet seg d e -AO, som fører til diamagnetisme kompleks, hvoretter det syvende og åttende elektronet går til d g-undernivå: Åpenbart med en åtte-elektronkonfigurasjon forskjeller i struktur mellom komplekser med ligander svak Og sterke felt forsvinner. Okkupasjonen av orbitaler av det niende og tiende elektronet er heller ikke forskjellig for komplekser av begge typer: La oss gå tilbake til betraktningen av den elektroniske strukturen til de oktaedriske komplekse ionene 3+ og 3-. I henhold til plasseringen i spektrokjemisk serie, er ammoniakk NH3 en av ligandene sterkt felt
, og fluoridion F - - svakt felt
. Følgelig vil okkupasjonen av atomorbitaler av elektroner i disse kompleksene skje i henhold til følgende skjema: I 3-anionet skaper F - liganden et svakt krystallfelt (D 0 = 13000 cm - 1), og alle elektronene til de opprinnelige 3 d 6 -JSC er plassert på d e - og d g orbitaler uten noen paring. Et komplekst ion er høyspinn og inneholder fire uparrede elektroner, så det paramagnetisk. I 3+-ionet skaper NH 3-ligander et sterkt krystallfelt (D 0 = 22900 cm - 1), alle 3 d 6 -elektroner er plassert på en mer energisk gunstig d e-orbitaler. Overføring av elektroner fra d e - på d g-orbitaler umulig fordi også høy energibarriere. Derfor er denne komplekse kationen lavt spinn, den inneholder ikke uparrede elektroner og diamagnetisk. På lignende måte kan skjemaer for fordeling av elektroner over orbitaler i et oktaedrisk felt for 2+ og 4-ioner presenteres: H 2 O-ligander skaper et svakt felt; utveksling av elektroner mellom d e - og d g-orbitaler forårsaker ingen vanskeligheter, og derfor er antallet uparrede elektroner i det komplekse ionet det samme som i det konvensjonelle Fe + II-ionet. Det resulterende vannkomplekset er høyt spinn, paramagnetisk. Mange komplekse forbindelser i krystallinsk tilstand og vandig løsning er sterkt farget. Således er en vandig løsning som inneholder 2+ kationer farget intenst blå, 3+ kationer gir løsningen en lilla farge, og 2+ kationer gir den en rød farge. Krystallfeltteorien gjør det mulig å forklare utseendet til en eller annen farge i komplekse forbindelser. Hvis lys sendes gjennom en løsning eller krystallinsk prøve av et stoff synlig del av spekteret, da er i prinsippet tre alternativer for den fysiske oppførselen til prøven mulig: ingen lysabsorpsjon hvilken som helst bølgelengde (stoffprøve fargeløs, selv om det kan ha absorpsjonsbånd i det ultrafiolette området av spekteret); fullstendig lysabsorpsjon over hele bølgelengdeområdet (prøven vil vises svart); endelig, lysabsorpsjon bare viss bølgelengde(da vil prøven ha farge komplementær til absorbert smal del av spekteret). Dermed bestemmes fargen på løsningen eller krystallene frekvens av absorpsjonsbånd synlig lys: Absorpsjonen av lyskvanter av komplekser (for eksempel de med en oktaedrisk struktur) forklares av samspillet mellom lys og elektroner lokalisert på d e -undernivå, ledsaget av deres overgang til ledige orbitaler d g-undernivå. For eksempel, når lys passerer gjennom en vandig løsning som inneholder heksakvatitan(III) 3+ kationer, detekteres et lysabsorpsjonsbånd i det gulgrønne området av spekteret (20300 cm - 1, l » 500 nm). Dette skyldes overgangen til enkeltelektronet til kompleksdannende middel fra d e-AO på d g-undernivå: Derfor får en løsning som inneholder 3+ en fiolett farge (i tillegg til den absorberte gulgrønne). En løsning av vanadiumsalt Cl 3 er grønn. Dette skyldes også de tilsvarende overgangene av elektroner når de absorberer en del av energien til lysstrålen. I grunntilstand, med den elektroniske konfigurasjonen av vanadium(III) 3 d 2 er to uparrede elektroner opptatt d e-undernivå: Det er kun to alternativer for overgang av to elektroner på d g -undernivå: enten både elektroner er okkupert d g -AO, eller bare en av dem. Alle andre elektronoverganger forbundet med en reduksjon i det totale spinnet er forbudt. Hvis kompleksdanneren har en elektronisk konfigurasjon d 0 eller d 10 da elektronoverganger Med d e - på d g -undernivå eller omvendt umulig enten fordi fravær av elektroner, enten fordi fravær av ledige orbitaler. Derfor absorberer ikke løsninger av komplekser med slike kompleksdannende midler som Sc(III), Cu(I), Zn(II), Cd(II), etc. energi i den synlige delen av spekteret og vises fargeløs: Selektiviteten til lysabsorpsjon avhenger ikke bare av kompleksdannende middel Og dens oksidasjonstilstand, men også fra type ligander. Når man erstatter ligander på venstre side av den spektrokjemiske serien i en kompleks forbindelse med ligander som skaper sterk elektrostatisk felt observert øke brøkdelen av energi som absorberes av elektroner fra transmittert lys og som en konsekvens, avta bølgelengden til det tilsvarende absorpsjonsbåndet. Således er en vandig løsning som inneholder tetraaquacopper(II) 2+ kationer blå, og en løsning av tetraamminecopper(II) 2+ sulfat er intens blå. ________________________ Gjenta: >>> applikasjoner
Energigevinst pga prioritert oppgjør elektroner d e-atomiske orbitaler kalles energi for stabilisering av komplekset av ligandfeltet.
lysets hastighet Med
= 3 ´ 10 10 cm/s.
Enhet D 0 er den samme som for bølgetallet n: cm - 1, som omtrent tilsvarer 12 J/mol.
I komplekse forbindelser som inkluderer kompleksdannende midler fra samme periode og i samme oksidasjonstilstand, med samme ligander, er spaltningsparameteren omtrent den samme. Med økende grad av oksidasjon av kompleksdanneren, verdien av D 0 øker. For vannkomplekser 2+ og 2+ er verdien av splitteparameteren 7800 og 10400 cm - 1, og for 3+ og 3+ - henholdsvis 13700 og 21000 cm - 1.
I-Br -Cl - » NCS - NEI 3 -F -ÅH - H2O » H - NH 3 NR 2 -CN - "NEI" CO. 









Omvendt forårsaker CN - ligander betydelig spaltning d-AO, som utgjør 33000 cm - 1. Dette betyr at det er en sterk tendens til å allokere alle elektroner på d e-orbitaler. Energigevinst, oppnådd med en slik populasjon av orbitaler, er mye større enn energikostnadene på grunn av elektronparing.

De indikerte overgangene til elektroner som har mottatt overskuddsenergi tilsvarer absorpsjonsbånd ca. 400 nm i absorpsjonsspekteret til en løsning av heksakvavanadium(III)klorid. Absorpsjon av det lilla-fiolette området av spekteret gir en ekstra farge til løsningen - lysegrønn.

Og John Van Vleck for å beskrive de lavere tilstandene til overgangsmetallkationer omgitt av ligander - både anioner og nøytrale molekyler. Krystallfeltteori ble ytterligere kombinert [og foredlet] med (delokalisert) molekylær orbitalteori til en mer generell teori som tar hensyn til den delvise kovalensen til metall-ligandbindingen i koordinasjonsforbindelser.
Krystallfeltteori lar en forutsi eller tolke de optiske absorpsjonsspektrene og elektronparamagnetiske resonansspektrene til krystaller og komplekse forbindelser, så vel som entalpiene for hydratisering og stabilitet i løsninger av overgangsmetallkomplekser.
Gjennomgang av Crystal Field Theory[ | ]
I følge TCP oppstår interaksjonen mellom et overgangsmetall og ligander fra tiltrekningen mellom det positivt ladede metallkationen og den negative ladningen av elektroner i ligandens ikke-bindende orbitaler. Teorien vurderer endringen i energi av fem degenerert d-orbitaler omgitt av punktladninger av ligander. Når liganden nærmer seg metallionet, blir ligandens elektroner nærmere noen d-orbitaler enn andre, forårsaker tap av degenerasjon. Elektroner d-orbitaler og ligander frastøter hverandre som ladninger med samme fortegn. Dermed energien til de d-elektroner som er nærmere ligandene blir høyere enn de som er lenger unna, noe som fører til en splittelse av energinivået d-orbitaler.
Følgende faktorer påvirker splittelsen:
- Naturen til metallionet.
- Graden av oksidasjon av metallet. Jo høyere oksidasjonstilstand, jo høyere er spaltningsenergien.
- Arrangement av ligander rundt et metallion.
- Naturen til liganden som omgir metallionet. Jo sterkere effekten av liganden er, desto større er forskjellen mellom høye og lave energinivåer.
Den vanligste typen ligandkoordinering er oktaedrisk, der seks ligander skaper et krystallfelt med oktaedrisk symmetri rundt metallionet. I det oktaedriske miljøet til et metallion med ett elektron i det ytre skallet, er d-orbitalene delt inn i to grupper med forskjell i energinivåer Δ okt ( fisjonsenergi), mens energien til orbitalene dxy, dxz Og d yz vil være lavere enn d z 2 og d x 2 -y 2, siden orbitalene til den første gruppen er lokalisert lenger fra liganden og opplever mindre frastøting. De tre lavenergiorbitalene er betegnet som t 2g, og to med høy-like f.eks.
De nest vanligste er tetraedrisk komplekser der fire ligander danner et tetraeder rundt et metallion. I dette tilfellet d-orbitaler er også delt inn i to grupper med forskjell i energinivå Δ tetr. I motsetning til oktaedrisk koordinasjon vil orbitalene ha lav energi d z 2 og d x 2 -y 2, og høy - d xy , d xz Og d yz. I tillegg, siden elektronene til liganden ikke er direkte i retningen d-orbitaler, vil spaltningsenergien være lavere enn ved oktaedrisk koordinasjon. Ved å bruke TCP kan du også beskrive plano-kvadrat og andre geometrier av komplekser.
Forskjellen i energinivåer Δ mellom to eller flere grupper av orbitaler avhenger også av arten av liganden. Noen ligander forårsaker mindre spaltning enn andre, årsakene til dette er forklart. Spektrokjemisk serie- en eksperimentelt oppnådd liste over ligander, ordnet i stigende rekkefølge Δ:
Oksydasjonstilstanden til metallet påvirker også Δ. Et metall med en høyere oksidasjonstilstand tiltrekker ligander nærmere på grunn av en større ladningsforskjell. Ligander nærmere metallionet forårsaker mer spaltning.
Lav- og høyspinnkomplekser[ | ]
Ligander som forårsaker stor spaltning d-nivåer, som CN− og CO, kalles ligander sterkt felt. I komplekser med slike ligander er det ugunstig for elektroner å okkupere høyenergiorbitaler. Følgelig blir lavenergiorbitaler fullstendig fylt før høyenergiorbitaler begynner å fylles. Slike komplekser kalles lavt spinn. For eksempel er NO 2 - en høyfeltsligand som produserer stor splitting. Alle 5 d-elektronene til det oktaedriske ionet 3− vil være plassert på lavere nivå t 2g .
Derimot kalles ligander som forårsaker liten spaltning, som I− og Br−, ligander svakt felt. I dette tilfellet er det lettere å plassere elektroner i høyenergiorbitaler enn å plassere to elektroner i samme lavenergiorbital, fordi to elektroner i samme orbital frastøter hverandre, og energikostnaden ved å plassere et andre elektron i orbitalen er høyere enn Δ. Således, før sammenkoblede elektroner dukker opp, i hver av de fem d-orbitaler må plasseres ett elektron om gangen i henhold til Hunds regel. Slike komplekser kalles høyspinn. For eksempel er Br− en svakfeltligand som forårsaker liten splittelse. Alle 5 d-orbitaler til 3−-ionet, som også har 5 d-elektroner vil være okkupert av ett elektron.
Splittingsenergien for tetraedriske komplekser Δ tetr er omtrent lik 4/9Δ okt (for samme metall og ligander). Som et resultat, forskjellen i energinivåer d-orbitaler er vanligvis under elektronparingsenergien, og tetraedriske komplekser er vanligvis høyspinn.
Distribusjonsdiagrammer d-elektroner gjør det mulig å forutsi de magnetiske egenskapene til koordinasjonsforbindelser. Komplekser med uparrede elektroner er paramagnetiske og tiltrekkes av et magnetfelt, mens komplekser uten er diamagnetiske og frastøter svakt.
Krystallfeltstabiliseringsenergi[ | ]
Krystallfeltstabiliseringsenergi (CFE) er energien til den elektroniske konfigurasjonen av et overgangsmetallion i forhold til den gjennomsnittlige energien til orbitalene. Stabilisering oppstår på grunn av det faktum at i ligandfeltet er energinivået til noen orbitaler lavere enn i et hypotetisk sfærisk felt, der alle fem d-orbitaler har samme frastøtende kraft, og alle d-orbitaler er degenererte. For eksempel, i oktaedrisk tilfelle nivået t 2g lavere enn gjennomsnittsnivået i et sfærisk felt. Følgelig, hvis disse orbitalene inneholder elektroner, er metallionet mer stabilt i ligandfeltet i forhold til det sfæriske feltet. Tvert imot energinivået til orbitalene f.eks over gjennomsnittet, og elektronene i dem reduserer stabiliseringen.
Stabiliseringsenergi av det oktaedriske feltet
Det er tre orbitaler i et oktaedrisk felt t 2g stabilisert i forhold til gjennomsnittlig energinivå med 2/5 Δ oktav, og de to orbitalene f.eks destabilisert med 3/5 Δ okt. Ovenfor var eksempler på to elektroniske konfigurasjoner d 5 . I det første eksemplet er det et lavspinnkompleks 3− med fem elektroner inn t 2g. Dens ESP er 5 × 2 / 5 Δ okt = 2Δ okt. I det andre eksemplet er et høyspinnkompleks 3− med ESKP (3 × 2 / 5 Δ okt) − (2 × 3 / 5 Δ okt) = 0. I dette tilfellet er den stabiliserende effekten av elektroner i lavnivåorbitaler nøytraliseres av den destabiliserende effekten av elektroner i orbitaler på høyt nivå.
Diagrammer over d-nivådeling av et krystallfelt[ | ]
| oktaedrisk | femkantet-bipyramidal | firkantet-antiprismatisk |
|---|---|---|
Krystallfeltteori erstattet teorien om valensbindinger på 40-tallet av XX-tallet. I sin rene form brukes den ikke for tiden, siden den ikke kan forklare dannelsen av kovalente bindinger i komplekse forbindelser og ikke tar hensyn til den sanne tilstanden til liganden (for eksempel deres faktiske størrelser) selv i tilfelle av nære interaksjoner til rent elektrostatisk.
Allerede på midten av 50-tallet ble den forenklede krystallfeltteorien erstattet av en forbedret ligandfeltteori, tar hensyn til den kovalente naturen til de kjemiske bindingene mellom kompleksdannende middel og liganden.
Imidlertid er den mest generelle tilnærmingen til å forklare dannelsen av komplekse forbindelser gitt av molekylær orbital teori(MO), som for tiden råder over alle andre. Den molekylære orbitalmetoden sørger for både ren elektrostatisk interaksjon i fravær av overlappende atomorbitaler, og hele settet med mellomliggende grader av overlapping.
La oss se på de grunnleggende konseptene krystallfeltteori, som i likhet med teorien om valensbindinger fortsatt beholder sin betydning for den kvalitative beskrivelsen av kjemiske bindinger i komplekse forbindelser på grunn av sin store enkelhet og klarhet.
I krystallfeltteori vurderes den kjemiske bindingen mellom kompleksdannende middel og liganden elektrostatisk. I følge denne teorien er ligandene lokalisert rundt det kompleksdannende middelet ved toppunktene til vanlige polyedre ( polyedre) som punktgebyrer. Teorien tar ikke hensyn til det faktiske volumet av liganden.
Ligander, som punktladninger, skaper rundt kompleksdannende middel elektrostatisk felt("krystallfelt", hvis vi vurderer en krystall av en kompleks forbindelse, eller ligandfelt), der energinivåene til kompleksdanneren og fremfor alt, d-undernivåer deler seg, og energien deres endres. Arten av splitting, energien til nye energinivåer avhenger av symmetri arrangement av ligander (oktaedrisk, tetraedrisk eller annet krystallfelt). Når molekylene H 2 O, NH 3 , CO og andre er koordinert som ligander, anses de som dipoler, orientert med en negativ ladning mot kompleksdanneren.
Tenk på tilfellet med et oktaedrisk arrangement av ligander (for eksempel -3 eller 3+). I midten av oktaederet er det et kompleksdannende ion M(+ n) med elektroner på d-atomiske orbitaler, og på toppene er det ligander i form av punkt negative ladninger (for eksempel F - ioner eller polare molekyler som NH 3). I et konvensjonelt ion M(+ n), ikke assosiert med ligander, er energiene til alle fem d-AO er de samme (dvs. atomorbitaler degenerert).
Imidlertid i det oktaedriske feltet av ligander d-AOs av kompleksdanneren faller inn i ulik posisjon. Atomiske orbitaler d(z 2) og d(x 2 -y 2), langstrakt langs koordinataksene, kommer nærmest ligandene. Mellom disse orbitalene og ligandene lokalisert ved toppunktene til oktaederet, oppstår det betydelige forskjeller frastøtende krefter, som fører til en økning i orbital energi. Disse atomorbitalene er med andre ord underlagt maksimal eksponering for ligandfeltet. En sterkt komprimert fjær kan tjene som en fysisk modell for slik interaksjon.
Tre andre d-AO - d(xy), d(xz) Og d(yz), plassert mellom koordinataksene og mellom liganden, er i større avstand fra dem. Samspillet mellom slike d-AO med ligander er minimal, og derfor energi d(xy), d(xz) Og d(yz)-AO reduseres sammenlignet med den opprinnelige.
Dermed femdoblet degenerert d-AO kompleksdannende middel, kommer inn oktaedrisk ligandfelt, utsatt splitting i to grupper av nye orbitaler – tre ganger degenererte orbitaler med lavere energi, d(xy), d(xz) Og d(yz), Og dobbelt degenererte orbitaler med høyere energi d(z 2) og d(x 2 -y 2). Disse nye gruppene d-orbitaler med Nedre Og høyere energi betegne dε og dγ:
d(z 2) og d(x 2 -y 2)
d(xy), d(xz),d(yz)
Energiforskjell to nye undernivåer dε og dγ ble navngitt deleparameter Δ 0:
E 2 – E 1 = Δ 0 ≈ 0
Plassering av to nye energi subnivåer dε og dγ i forhold til originalen ( d-AO) på energidiagrammet asymmetrisk:
(E 2 – E 0) > (E 0 – E 1).
Kvantemekanisk teori krever at når nye energinivåer er fullstendig befolket med elektroner, forblir den totale energien uendret, dvs. hun burde bli lik E 0 .
Likheten skal med andre ord tilfredsstilles
4(E 2 – E 0) = 6(E 0 – E 1),
hvor 4 og 6 – maksimum antall elektroner pr dγ- og d e-AO. Av denne likestillingen følger det at
(E 2 – E 0) / (E 0 – E 1) = 3/2 og
(E 2 – E 1) / (E 0 – E 1) = 5/2, eller
Δ 0 / ( E 0 – E 1) = 5/2, hvorfra ( E 0 – E 1) = 2/5Δ0.
Plasser hvert elektron ut av de maksimale seks mulige på dε orbitaler årsak avta (gevinster) energi med 2/5 Δ0.
Tvert imot, plasseringen av hvert elektron av fire mulig på dγ orbitaler årsak øke (koste) energi med 3/5 Δ 0 .
Hvis befolket med elektroner dε- og dγ-orbitaler fullstendig, så nei vinne energi vil ikke(akkurat som det ikke vil ekstra energiforbruk).
Men hvis originalen d-AO er kun befolket delvis og inneholder fra 1 til 6 elektroner, og disse elektronene plasseres kun på dε-AO, da får vi betydelig energigevinst.
Energigevinst pga prioritert oppgjør elektroner dε-atomorbitaler kalles energi for stabilisering av komplekset av ligandfeltet.
Spesifisiteten til hver ligand påvirker feltet som denne liganden skaper - sterk eller svak. Hvordan sterkere felt ligander enn mer betydning deleparameter Δ 0 .
Studiet av splittingsparameteren er vanligvis basert på spektroskopisk forskning. Bølgelengder absorpsjonsbånd komplekser i krystallinsk tilstand eller i løsning, på grunn av overføring av elektroner fra dε- på dγ-AO, assosiert med deleparameterΔ 0 som følger:
λ = c / ν; Δ 0 = E 2 – E 1 = h ν = h · ( c / λ),
hvor er Plancks konstant h lik 6,6260693 ∙ 10 -34 J s;
lysets hastighet Med
= 3 · 10 10 cm/s.
EnhetΔ 0 er det samme som bølgetallet: cm -1, som omtrent tilsvarer 12 J/mol. Delingsparameter, i tillegg til typen ligand, avhenger på graden av oksidasjon Og natur kompleksdannende middel.
I komplekse forbindelser som inkluderer kompleksdannende midler fra samme periode og i samme oksidasjonstilstand, med samme ligander, er spaltningsparameteren omtrent den samme. Med økende grad av oksidasjon av kompleksdannende middel er verdien Δ 0 øker. For vannkomplekser 2+ og 2+ er verdien av spaltningsparameteren 7800 og 10400 cm -1, og for henholdsvis 3+ og +3 13700 og 21000 cm -1. På økende atomladning av det kompleksdannende atomet Δ 0 øker også. Hexaamminiridium(III) 3+, hexaamminiridium(III) 3+ og hexaamminiridium(III) 3+ kationer ( Z= 27, 45 og 77) er karakterisert ved å dele parametere lik 22900, 34100 og 41000 cm -1.
Avhengigheten av Δ 0 av naturen til liganden er mer mangfoldig. Som et resultat av studiet av en rekke komplekse forbindelser, ble det funnet at når det gjelder deres evne til å øke spaltningsparameteren til kompleksdannende metaller lokalisert i deres vanlige oksidasjonstilstander, kan de vanligste ligander ordnes i følgende spektrokjemisk serie, langs hvilken verdien Δ 0 øker monotont:
I > Br > Cl > NCS - ≈ NO 3 - > F - > OH - > H 2 O > H - > NH 3 > NO 2 - > CN - > NO > CO.
Dermed det sterkeste elektrostatiske feltet rundt kompleksdanneren og den sterkeste spaltningen d-AO er forårsaket av liganden CN-, NO og CO. La oss vurdere fordelingen av elektroner over dε- og dγ-orbitaler i det oktaedriske feltet til ligander. Innsjekking dε- og dγ-orbitaler oppstår i full overensstemmelse med Hunds regel Og Pauli-prinsippet. I dette tilfellet, uavhengig av verdien av splittingsparameteren, er de tre første elektronene okkupert av kvanteceller dε-undernivå:
![]() dε
dε
Dersom antall elektroner pr d- det er mer enn tre undernivåer av kompleksdanneren; det er to muligheter for å plassere dem på delte undernivåer. Ved en lav verdi av spaltningsparameteren (svake felt av liganden), overvinner elektroner energibarrieren. dε- og d y-orbitaler; det fjerde og deretter det femte elektronet befolker kvanteceller dγ-undernivå.
![]() dε
dε
Med et sterkt ligandfelt og en høy verdi på Δ 0, populasjonen av det fjerde og femte elektronet dγ-undernivå er ekskludert; utfylling pågår dε-orbitaler.
![]() dε
dε
På svake feltligander fylle kvanteceller med 4 eller 5 elektroner parallelle spinn, så det resulterende komplekset viser seg å være sterkt paramagnetisk. I et sterkt ligandfelt ett og deretter to elektronpar dannes på dε-undernivå, altså paramagnetisme komplekset viser seg å være mye svakere. Det sjette, syvende og åttende elektronet i tilfelle av et svakt felt ender opp igjen dγ-undernivå, som komplementerer konfigurasjonene til elektronpar (ett i tilfellet d 6, to – d 7 og tre - d 8):
![]() dε
dε
Ved et sterkt ligandfelt fyller det sjette elektronet seg dε-AO, fører til diamagnetisme kompleks, hvoretter det syvende og åttende elektronet går til dγ-undernivå:
![]() dε
dε
Åpenbart med en åtte-elektronkonfigurasjon forskjeller i struktur mellom komplekser med ligander svak Og sterke felt forsvinner. Okkupasjonen av orbitaler av det niende og tiende elektronet er heller ikke forskjellig for komplekser av begge typer:
![]() dε
dε
La oss gå tilbake til betraktningen av den elektroniske strukturen til de oktaedriske komplekse ionene 3+ og -3. I henhold til plasseringen i spektrokjemisk serie, er ammoniakk NH3 en av ligandene sterkt felt, og fluoridion F - - svakt felt. I -3 anion skaper F - liganden et svakt krystallfelt (Δ 0 = 13000 cm -1), og alle elektronene til de opprinnelige 3 d 6 -JSC er plassert på dε- og dγ orbitaler uten noen paring. Et komplekst ion er høyspinn og inneholder fire uparrede elektroner, så det paramagnetisk:

I 3+-ionet skaper NH 3-ligander et sterkt krystallfelt (Δ 0 = 22900 cm -1), alle 3 d 6 -elektroner er plassert på en mer energisk gunstig dε-orbitaler. Overføring av elektroner fra dε- på dγ orbitaler umulig fordi også høy energibarriere. Derfor er denne komplekse kationen lavt spinn, den inneholder ikke uparrede elektroner og diamagnetisk:

På lignende måte kan skjemaer for fordeling av elektroner over orbitaler i et oktaedrisk felt for 2+ og -4 ioner presenteres:


H 2 O-ligander skaper et svakt felt; utveksling av elektroner mellom dε- og dγ-orbitaler forårsaker ingen vanskeligheter og derfor er antallet uparrede elektroner i det komplekse ion det samme som i det konvensjonelle ionet Fe + II. Det resulterende vannkomplekset er høyt spinn, paramagnetisk.
Omvendt forårsaker CN - ligander betydelig spaltning d-AO, som utgjør 33000 cm -1. Dette betyr at det er en sterk tendens til å allokere alle elektroner på dε-orbitaler. Energigevinst, oppnådd med en slik populasjon av orbitaler, er mye større enn energikostnadene på grunn av elektronparing.
Fra perspektivet til valensbindingsmetoden innebærer hybridisering av valensorbitaler som danner en binding i vannkomplekset d-AO eksternt undernivå (4 sp 3 d 2), og i lavspinn - d-JSC internt undernivå (3 d 2 4sp 3).
Således, i høyspinnkomplekser med svakfeltsligander, forekommer hybridisering med deltakelse d-AO av det ytre undernivået, og lavspinn med høyfeltsligander - d- JSC for det interne undernivået. Antall uparrede elektroner i komplekset kan bestemmes ved elektronparamagnetisk resonans (EPR). Ved å bruke instrumenter av denne metoden, kalt EPR-spektrometre, studeres paramagnetiske stoffer.
Krystallfeltteorien gjør det mulig å forklare utseendet til en eller annen farge i komplekse forbindelser. Blant komplekse forbindelser kjennetegnes et betydelig antall i krystallinsk tilstand og vandig løsning ved deres lyse farge. Således er en vandig løsning som inneholder 2+ kationer farget intenst blå, 3+ kationer gir løsningen en fiolett farge, og 2+ kationer gir den en rød farge. Hvis lys sendes gjennom en løsning eller krystallinsk prøve av et stoff synlig del av spekteret, da er i prinsippet tre alternativer for den fysiske oppførselen til prøven mulig: ingen lysabsorpsjon hvilken som helst bølgelengde (stoffprøve fargeløs, selv om det kan ha absorpsjonsbånd i det ultrafiolette området av spekteret); fullstendig lysabsorpsjon over hele bølgelengdeområdet (prøven vil vises svart); endelig, lysabsorpsjon bare viss bølgelengde(da vil prøven ha farge komplementær til absorbert smal del av spekteret).

Dermed bestemmes fargen på løsningen eller krystallene frekvens av absorpsjonsbånd synlig lys. Absorpsjonen av lyskvanter av komplekser (for eksempel de med en oktaedrisk struktur) forklares av samspillet mellom lys og elektroner lokalisert på dε-undernivå, ledsaget av deres overgang til ledige orbitaler dγ-undernivå. For eksempel, når lys passerer gjennom en vandig løsning som inneholder heksakvatitan(III) 3+ kationer, detekteres et lysabsorpsjonsbånd i det gulgrønne området av spekteret (20300 cm -1, λ=500 nm). Dette skyldes overgangen til enkeltelektronet til kompleksdannende middel fra dε-AO på dγ-undernivå:

Derfor får en løsning som inneholder 3+ en fiolett farge (i tillegg til den absorberte gulgrønne). En løsning av vanadiumsalt Cl 3 er grønn. Dette skyldes også de tilsvarende overgangene av elektroner når de absorberer en del av energien til lysstrålen. I grunntilstand, med den elektroniske konfigurasjonen av vanadium(III) 3 d 2 er to uparrede elektroner opptatt dε-undernivå:

Det er kun to alternativer for overgang av to elektroner på dγ-undernivå: enten både elektroner er okkupert dγ-AO, eller bare en av dem. Alle andre elektronoverganger forbundet med en reduksjon i det totale spinnet er forbudt.
De indikerte overgangene til elektroner som har mottatt overskuddsenergi tilsvarer absorpsjonsbånd ca. 400 nm i absorpsjonsspekteret til en løsning av heksakvavanadium(III)klorid. Absorpsjon av det lilla-fiolette området av spekteret gir en ekstra farge til løsningen - lysegrønn. Hvis kompleksdanneren har en elektronisk konfigurasjon d 0 eller d 10 da elektronoverganger Med dε- på dγ-undernivå eller omvendt umulig enten fordi fravær av elektroner, enten fordi fravær av ledige orbitaler. Derfor er løsninger av komplekser med kompleksdannende midler som Sc(III) (3 d 0), Cu(I) (3 d 10), Zn(II) (3 d 10), Cd(II) (4 d 10), etc., absorberer ikke energi i den synlige delen av spekteret og vises fargeløs. Selektiviteten til lysabsorpsjon avhenger ikke bare av kompleksdannende middel Og dens oksidasjonstilstand, men også fra type ligander. Når man erstatter ligander på venstre side av den spektrokjemiske serien i en kompleks forbindelse med ligander som skaper sterk elektrostatisk felt observert øke brøkdelen av energi som absorberes av elektroner fra transmittert lys og som en konsekvens, avta bølgelengden til det tilsvarende absorpsjonsbåndet. Således er en vandig løsning som inneholder tetraaquacopper(II) 2+ kationer blå, og en løsning av tetraamminecopper(II) 2+ sulfat er intens blå.
Relatert informasjon.
