Понятие о теории кристаллического поля. Модели химической связи. Теория кристаллического поля. Низко- и высокоспиновые комплексы
Слабое поле сильное поле
Среднее поле
Frac34;¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾¾® Δo
Лиганды слабого поля с элементами 3d-ряда образуют высокоспиновые комплексы, а лиганды сильного поля – низкоспиновые. Различие между ними сказывается на электронном строении комплексов лишь для конфигураций d 4 – d 7:
3+ d 5 3– d 5
высокоспиновый комплекс низкоспиновый комплекс
H 2 O – лиганд слабого поля CN – – лиганд сильного поля
Низкоспиновые комплексы всегда устойчивее высокоспиновых. Лиганды среднего поля в зависимости от условий (заряд и природа центрального атома) могут образовывать как высокоспиновые, так и низкоспиновые комплексы.
Пример. На основании ТКП выскажите предположение об электронном строении ионов гексаамминкобальта(II) (Δo = 21600 см –1 , Р = 21000 см –1) и гексаамминкобальта(III) (Δo = 9500 см –1 , Р = 22500 см –1).
Аммиак – лиганд среднего поля и в зависимости от степени окисления металла может образовывать как высокоспиновые, так и низкоспиновые комплексы. Выясним, какие комплексы будут энергетически более устойчивы для кобальта(II) и кобальта (III). Для этого сравним ЭСКП каждого из ионов в сильном и слабом поле:
(а) 3+ , d 6

сильное поле слабое поле
ЭСКП (сильное поле) = –6´(2/5)Δo + 2P = –6´(2/5) ´21600 + 2´21000 = –9840 см –1
ЭСКП (слабое поле) = –4´(2/5)Δo + 2´(3/5)Δo = –4´(2/5) ´21600 + 2´(3/5) ´21600 = –8640 см –1
Энергетический выигрыш больше в случае низкоспинового комплекса.
(б) 2+ , d 7

сильное поле слабое поле
ЭСКП (сильное поле) = –6´(2/5)Δo + 1´(3/5)Δo + P = –6´(2/5)´9500 + 1´(3/5) ´9500 + 22500 = 7900 см –1
ЭСКП (слабое поле) = –5´(2/5)Δo + 2´(3/5)Δo = –5´(2/5) ´9500 + 2´(3/5) ´9500 = –7600 см –1
Энергетический выигрыш больше в случае высокоспинового комплекса.
Таким образом, ион 3+ является низкоспиновым, а 2+ - высокоспиновым.
ЭСКП возрастает с увеличением Δo, однако, различно для высокоспинового и низкоспинового состояний (Рис. 1.28. Зависимость ЭСКП для высокоспинового и низкоспинового комплексов с конфигурацией d 6 от величины Δo = 10Dq. Область, в которой возможно существование обоих состояний, заштрихована). Область вблизи точки пересечения этих двух прямых соответствует комплексам, способным существовать как в высокоспиновом, так и в низкоспиновом состояниях.
Примером может служить тиоцианатный комплекс железа(II) с 1,10-фенантролином , который при низких температурах высокоспиновый (парамагнитный), а при повышении температуры – низкоспиновый (диамагнитный) (M. Marchivie, P. Guionneau, J. A. K. Howard, G. Chastanet, J.-F. Letard, A. E. Goeta, D. Chasseau, J. Am. Chem. Soc., 2002, v. 124, p. 194). Изменение мультиплетности сопровождается изменением межатомных расстояний и геометрии координационного окружения: низкоспиновый комплекс представляет собой правильный октаэдр, а высокоспиновый – искаженный. Обратный переход в высокоспиновое состояние возможен под действием высоких давлений или излучения. В настоящее время известно несколько десятков подобных систем.
Говоря о σ-донорных и π-акцепторных свойствах лиганда, мы вышли за рамки ТКП, используя подходы метода молекулярных орбиталей применительно к комплексным соединениям (том 1). Напомним, что картина расщепления d-орбиталей является фрагментом общей схемы молекулярных орбиталей в октаэдрическом комплексе, где t 2g -орбитали рассматриваются как несвязывающие, а e g – как разрыхляющие (рис. том 1).
В образовании связей в октаэдрическом комплексе без π-связывания участвуют s-, p- и d-орбитали металла и по одной орбитали от каждого лиганда. Из 15 атомных орбиталей образуется 15 молекулярных, шесть из них (a 1 g , t 1 u , e g (сноска: буква в обозначении орбиталей указывает на степень их вырожденности: t – трижды вырожденные, e – дважды вырожденные, a – невырожденные, и на наличие центра симметрии: g – симметричные, u - несимметричные)) σ-связывающие, три (t 2 g) – несвязывающие, и шесть (e g *, t 1 u *, a 1 g *) σ-разрыхляющие. Связывающие орбитали по энергии ближе к орбиталям лиганда, а несвязывающие локализованы преимущественно на атоме металла. Энергия d xy , d xz , d yz (t 2 g) орбиталей металла при образовании комплекса практически не изменяется.
Наличие у лиганда низкой по энергии вакантной орбитали, сходной по симметрии с орбиталями металла, приводит к понижению энегии t 2g -орбиталей, практически не влияя на e g , тем самым увеличивая Δо (Рис. 1.29. Фрагменты диаграммы МО для комплекса кобальта(III) с σ-донорным лигандом (а) и σ-донорным, π-акцепторным лигандом (б)).
Эффект Яна-Теллера. В 1937 г. Ян и Теллер доказали теорему, согласно которой любая нелинейная молекула в вырожденном электронном состоянии неустойчива и самопроизвольно претерпевает искажение, понижающее ее симметрию и приводящее к снятию вырождения. Теорема предсказывает лишь сам факт снятия вырождения, но не указывает, каким образом оно будет снято. На основании этой теоремы получило объяснение искажение октаэдрической геометрии ряда комплексов, а сам факт наличия такого искажения получил название эффекта Яна-Теллера. Обратимся к примеру. Комплексы меди(II) с конфигурацией d 9 , как правило, не представляют собой правильный октаэдр, а вытянуты или сжаты по одной из осей (Рис. 1.30. Искажение октаэдрической геометрии в комплексах меди (II)). Рассмотрим случай вытянутого октаэдра. Удаление лигандов, расположенных по оси z, вызывает снятие вырождения вследствие изменения энергий орбиталей. Орбитали, направленные по оси z (d xz , d yz , d z 2), слабее взаимодействуют с орбиталями лигандов по сравнению с орбиталями, не имеющими z-компоненты (d xy , d x 2 -y 2), и поэтому понижают свою энергию. Пара орбиталей одинаковой симметрии, имеющие z-компоненту (d xz , d yz), остается вырожденной и приобретает повышенную энергию. (Рис. 1.31. Изменение энергий d-орбиталей при искажении октаэдра). Эффект Яна-Теллера с наибольшей силой проявляется в комплексах с неравноценно заполненными e g -орбиталями, то есть с конфигурациями t 2g 3 e g 1 (соответствует иону d 4 в слабом поле: CrCl 2 , K 3 MnF 6) и t 2g 6 e g 3 (соответствует иону d 9: практически все комплексы меди(II)) и t 2g 6 e g 1 (соответствует иону d 7 в сильном поле, встречается редко, K 3 NiF 6),. Незначительный эффект Яна-Теллера характерен для комплексов с неравноценно заполненными t 2g -орбиталями, то есть для электронных конфигураций t 2g 1 (d 1), t 2g 2 (d 2), t 2g 4 (d 4 в сильном поле), t 2g 5 (d 5 в сильном поле), t 2g 5 e g 1 (d 6 в слабом поле), t 2g 5 e g 2 (d 7 в слабом поле). Ионы с конфигурациями d 3 и d 5 в слабом поле, d 3 и d 6 в сильном поле, d 8 и d 10 ни при каких условиях не являются ян-теллеровскими.
Эффект Яна-Теллера проявляется в неравноценности длин связей во многих комплексах меди(II) и марганца(III), в немонотонном изменении ступенчатых констант устойчивости комплексов. Например, в безводном хлориде меди(II) атом меди расположен в окружении из шести атомов хлора, четыре из которых находятся на расстоянии 0,230 нм, а два других удалены от него на 0,295 нм.
Известны комплексы меди(II) (Cl 2 , (C 6 H 5 SO 3) 2 и др), состоящие из нескольких кристаллографически неэквивалентных ян-теллеровских ионов, каждый со своим типом искажения, которые превращаются друг в друга, изменяя расстояние металл-лиганд настолько быстро, что в целом все расстояния металл-лиганд кажутся одинаковыми. Этот случай получил название динамического, или пульсирующего эффекта Яна-Теллера (P. E. M. Wijnands, J. S. Wood, J. Redijk, W. J. A. Maaskant, Inorg. Chem., 1986, 35, 1214) .
Эффект Яна-Теллера, тем не менее, не относится к всеобщим законам. В настоящее время известны комплексные ионы с ян-теллеровской конфигурацией, представляющие собой неискаженные октаэдры: 4– , 3+ .
Расщепление в полях с симметрией, отличной от октаэдрической .
Помимо октаэдрических, известно множество комплексов с иной геометрией – плоско-квадратных, тетраэдрических, тригонально-пирамидальных, квадратно-пирамидальных, линейных и др. Расщепление в каждом из этих полей иное, чем в октаэдре, оно определяется симметрией координационного полиэдра.
Плоско-квадратные комплексы можно рассматривать как предельный случай тетрагонального искажения октаэдрической геометрии, когда лиганды, расположенные по одной из координатных осей, удалены в бесконечность (Рис. 1.27б). Обозначения орбиталей приведены на рисунке. Плоско-квадратные комплексы наиболее типичны для ионов с электронной конфигурацией d 8 – Ni 2+ , Pd 2+ , Pt 2+ , Au 3+ . Их устойчивость резко возрастает при увеличении Δ, то есть при переходе от элементов 3d-ряда к тяжелым переходным элементам. Так, например, если у палладия, платины и золота практически все комплексы с координационным числом четыре квадратные, то никель образует плоско-квадратные комплексы лишь с лигандами сильного поля: 2– , Ni(dmg) 2 . Комплексы никеля(II) с лигандами слабого поля, например, с галогенами, имеют тетраэдрическую геометрию.
Некоторые плоско-квадратные комплексы переходных металлов в твердом виде образуют цепи с мостиковыми лигандами, например, Pt-CN-Pt в K 2 Br 0,3 , где атомы платины частично находятся в степени окисления +4. Высокая проникающая способность 5d-орбиталей обеспечивает их перекрывание с образованием единой энергетической зоны, а, следовательно, и металлическую проводимость по направлению цепи. Такие молекулярные комплексы способны проводить электрический ток, и в настоящее время интенсивно изучаются.
В поле тетраэдрической симметрии максимальной энергией обладают орбитали d xy , d yz , d xz , их называют t 2 -орбиталями, а минимальной – орбитали d x 2 –y 2 и d z 2, их обозначают e. Из-за наличия меньшего числа лигандов и иного их расположения тетраэдрическое поле (Рис. 1.32. Сравнение расщеплений в тетраэдрическом и октаэдрическом поле) оказывается в 2,25 раза слабее октаэдрического: .
Большинство тетраэдрических комплексов высокоспиновые.(Сноска – Известны несколько примеров низкоспиновых тетраэдрических комплексов, например, Cr{N(Si(CH 3) 3) 2 } 3 NO (хром(II), d 4 ; D. C. Bradley, Chem. Ber., 1979, 11, 393); CoL 4 , где L – 1-норборнил (кобальт(IV), d 5 ; E. K: Brune, D. S. Richeson, K. H. Theopold, Chem. Commun., 1986, 1491)). Максимальная стабилизация тетраэдрического окружения кристаллическим полем достигается при конфигурациях d 2 (FeO 4 2– , MnO 4 3–) и d 7 ( 2–). По причине относительно низкой энергии стабилизации тетраэдрические комплексы образуются чаще ионами с конфигурациями d 0 (TiCl 4 , MnO 4 – , CrO 4 2–), d 5 в слабом поле (FeCl 4 –) и d 10 (ZnCl 4 2–) с нулевой ЭСКП, а также ионами непереходных металлов (AlCl 4 –). Образованию тетраэдрических комплексов по сравнению с октаэдрическими часто благоприятствует стерический фактор, так, ион – более устойчивый, чем 3– .
Использование ТКП для объяснения устойчивости комплексов. Ряд Ирвинга-Уильямса. Теория кристаллического поля позволяет объяснить немонотонный характер изменения энергий кристаллической решетки оксидов и галогенидов, констант устойчивости комплексов и др. Порядок изменения энергий гидратации двухзарядных катионов 3d-металлов в целом совпадает с характером изменения ЭСКП в высокоспиновых комплексах (Рис. 1.33. Изменение энергии гидратации двухзарядных катионов металлов 3d-ряда (а) и изменение ЭСКП в высокоспиновых комплексах (б)), чем сильнее стабилизация кристаллическим полем, тем больше гидратация. Известно, что константы замещения молекулы воды на лиганд слабого поля L
2+ + L x– = (2-x)+ + H 2 O
подчиняются ряду Ирвинга-Уильямса: Mn 2+ < Fe 2+ < Co 2+ < Ni 2+ < Cu 2+ < Zn 2+ (Рис. 1.34. Зависимость первой константы устойчивости комплекса от природы 3d-металла). Согласно этому ряду, наибольшей устойчивостью обладают комплексы меди(II) и никеля(II). Простейший вариант ЭСКП предсказывает наибольшую устойчивость никелевых комплексов. При этом надо учитывать, что комплексы меди(II) имеют сильно искаженную октаэдрическую геометрию, что вносит существенный вклад в величину константы устойчивости.
Нефелоауксетический эффект. Обнаружено, что взаимное отталкивание d-электронов ослабевает при помещении атома в поле лигандов. Такое воздействие лиганда на d-электроны атома металла получило название нефелоауксетического эффекта от греческих слов νεφελη – облако и αυξανω – увеличивать. Ряд лигандов, расположенных в порядке усиления их воздействия на орбитали металла, практически полностью соответствует спектрохимическому ряду. Причиной нефелоаксетического эффекта служит перекрывание d-орбиталей металла с орбиталями лигандов, благодаря чему d-облако расширяется в пространстве. Наличие этого эффекта наглядно демонстрирует ограниченность простейшей элеткростатической модели – теории критсллического поля, предполагающей, что лигнады являются точечными отрицательными зярядами.
Теория поля лигандов. Теория кристаллического поля была разработана Бете (Bethe) в 1929 г. В настоящее время она находит широкое применение в модифицированном виде с поправками на некоторую долю ковалентности связи металл-лиганд. Такая теория называется теорией поля лигандов. Наличие ковалентного вклада изменяет энергию орбиталей металла по сравнению с рассчитанной по ТКП. Долю ковалентности учитывают введением поправочных коэффициентов, позволяющих приравнять экспериментальные значения к рассчитанным.
Окраска комплексов.
Окраска комплексов d-переходных элементов связана с переходами электронов с одной d-орбитали на другую. Это наглядно иллюстрирует пример иона Ti 3+ , рассмотренный в первом томе учебника. Поглощая энергию, соответствующую синей и зеленой части видимого спектра, единственный d-электрон в ионе Ti 3+ переходит на e g -орбиталь (Рис. 1.35. Спектр иона 3+). Окраска иона обусловлена дополнительными цветами – красным и фиолетовым. (Сноска – Внимательный читатель заметит некоторую асимметрию полосы поглощения. Она является следствием незначительного расщепления t 2g -уровня, вызванного эффектом Яна-Теллера). Диаграмма, на которой изображены дополнительные цвета и которая хорошо известна каждому художнику, представлена на втором форзаце учебника. Энергия перехода, выраженная в обратных сантиметрах (1000 см –1 = 12 кДж), соответствует параметру расщепления Δο – его чаще всего и определяют из электронных спектров. Длина волны обратно пропорциональна энергии:
 .
.
В случае комплексов с большим числом электронов картина спектра усложняется, в нем появляются дополнительные полосы. Это связано с тем, что возбужденное состояние t 2g 1 e g 1 может быть реализовано несколькими способами в зависимости от того, на каких двух d-орбиталях находятся электроны. Например, состояние, при котором электроны занимают d xy и d x 2 –y 2 орбитали, будет выше по энергии, чем состояние d xy 1 d z 2 1 , из-за большего отталкивания электронов по оси x. Энергия, соответствующая полосе с наименьшей энергией, равна параметру расщепления Δo.
Чтобы описать электронные спектры более детально, необходимо ввести некоторые понятия. Любое расположение электронов на подуровне назовем микросостоянием. Число микросостояний N, при которых n электронов занимают х орбиталей, равно

Каждое микросостояние характеризуется собственными значениями спинового и углового моментов. Набор микросостояний с одинаковыми энергиями называется термом , например, 3 Р, 5 D, 1 S. Цифровой индекс обозначает мультиплетность, которая рассчитывается как:
мультиплетность = число неспаренных электронов в основном состоянии + 1.
Названия термов читаются с указанием мультиплетности: “триплет Р”, “квинтет D”, “синглет S”. Буква обозначает суммарный угловой момент L атома или иона, который равен максимальному значению суммы угловых моментов m l отдельных орбиталей, занятых электронами. Например, ион Ti 3+ содержит один d-электрон, число микросостояний N = (2´5)!/1!(2´5 – 1)! = 10, L = 2(D) (т.к. для d-орбитали m l = –2, –1, 0, 1, 2, число электронов равно 1, следовательно, максимальная сумма m l равна наибольшему значению m l), мультиплетность 1 + 1 = 2. Следовательно, терм основного состояния (с наименьшей энергией) 2 D. В случае иона с электронной конфигурацией d 2 N = (2´5)!/2!(2´5 – 2)! = 45, L = 3(F) (т.к. для d-орбитали m l = –2, –1, 0, 1, 2, число электронов равно 2, следовательно, максимальная сумма двух наибольших значений равна m l), мультиплетность 2 + 1 = 3. Следовательно, терм основного микросостояния 3 F. При ином расположении двух электронов на d-подуровне достигаются состояния, описываемые другими термами – 3 P, 1 G, 1 D, 1 S и т.д. Соотношение между численными значениями L и буквенными символами приведено ниже:
L = 0 1 2 3 4 5 6 7
Аналогично можно вывести термы основных и возбужденных состояний и для других ионов d-элементов (Табл. 1.5.). Обратите внимание, что термы ионов с конфигурацией d n и d 10-n совпадают.
Таблица. 1.5.
Термы основного и ближайших возбужденных состояний для различных конфигураций d-электронов.
Термы расщепляются в октаэдрическом поле подобно орбиталям, обозначаемым аналогичными буквами. D термы расщепляются на T 2 g и E g составляющие, подобно d-орбиталям, F термы – на T 1 g , T 2 g и A 2 g , подобно f-орбиталям. S и Р термы вообще не расщепляются. Возможности перехода электронов между различными состояниями ограничиваются правилами отбора. Так, в комплексах разрешены лишь переходы между состояниями с одинаковой мультиплетностью. Каждому такому переходу соответствует полоса в спектре поглощения. В качестве примера рассмотрим электронный спектр комплекса 3+ (Рис. 1.36. Электронный спектр комплекса 3 +). Три полосы обусловлены тремя электронными переходами: 4 A 2 g ® 4 T 2 g , 4 A 2 g ® 4 T 1 g , 4 A 2 g ® 4 T 1 g (P). Переход с наименьшей энергией соответствует величине параметра расщепления: Δo = 17400 см –1 . Комплекс поглощает свет в красной (17400 см –1) и голубой (23000 см –1) частях видимого спектра и в ближнем ультрафиолете (37800 см –1), следовательно, он имеет фиолетовую окраску.
Согласно правилу Лапорта, переходы между состояниями с одинаковой четностью, к которым относятся s-s, p-p, d-d, f-f-переходы, маловероятны, или, на языке спектроскопии, в октаэдрических комплексах они запрещены. Запрещенные переходы возможны, но протекают с низкой интенсивностью. Именно поэтому соли переходных металлов имеют заметную окраску лишь в концентрированных растворах. Она во много раз слабее окраски перманганата или дихромата, в ионах которых не содержится d-электронов.
Правило Лапорта применимо лишь в случае комплексов, имеющих центр симметрии. При искажении октаэдра центр симметрии исчезает, запрет Лапорта снимается, и появляется окраска. Например, ион 3+ бесцветный, однако растворы солей железа(III) часто окрашены в желто-оранжевый цвет из-за гидролиза, приводящего к образованию несимметричных частиц с искаженным октаэдрическим окружением.
Окраску комплексов, помимо d-d переходов с одной d-орбитали металла на другую (с t 2g на e g в октаэдрических комплексах), обусловливают еще два фактора: переходы с орбителей лиганда на орбитали металла (их называют переносом заряда) и переходы внутри орбиталей лиганда. Эти переходы не подпадают под правило Лапорта, а, следовательно, имеют высокую интенсивность.
Полоса переноса заряда присутствует в электронном спектре любого соединения, однако, в ряде случаев она находится в ультрафиолетовой части спектра и не воспринимается нами как окраска. Если разность между энергиями орбиталей лиганда и орбиталей металла сокращается, полоса переноса заряда попадает в видимую часть спектра. Именно переносом заряда объясняется интенсивная окраска перманганата, дихромата, сульфида ртути, пероксокомплексов титана(IV) и многих других соединений с пустыми d-орбиталями. В ряде случаев под действием света перенос заряда с орбиталей лиганда на орбитали металла происходит необратимо, то есть сопровождается химическим процессом. Примером служит фотохимическое разложение галогенидов серебра, лежащее в основе черно-белой фотографии: Ag + Br – ¾® Ag 0 + Br 0 .
В электронном спектре перманганата калия наблюдаются четыре полосы, соответствующие переходам электронов с несвязывающих орбиталей, локализованных преимущественно на лиганде (a 1 , t 2 σ-орбитали и e, t 1 , t 2 π-орбитали), на e*, t2’’разрыхляющие орбитали, локализованные на атоме металла ((рис.1.37. Энергетическая диаграмма тетраэдрического иона MnO 4 - с π-связыванием. Переходы электронов показаны стрелками):
ν 1 , Mn(e*) ¾ O(t 1) 17700 см –1
ν 2 , Mn(t 2 ’’) ¾ O(t 1) 29500 см –1
ν 3 , Mn(e*) ¾ O(t 2) 30300 см –1
ν 4 , Mn(t 2 ’’) ¾ O(t 2) 44400 см –1
Полоса с наименьшей энергией попадает в видимую часть спектра (λ = 107/17700 = 565 нм), что соответствует поглощению зеленого света и пропусканию малиново-красного.
3. Механизмы реакций с участием комплексных соединений.
Подавляющее большинство химических процессов протекает как последовательная цепь некоторых элементарных стадий, а уравнение реакции несет лишь информацию о главных конечных продуктах реакции. Эта последовательность элементарных превращений на пути от исходных веществ к продуктам и называется механизмом. Промежуточные, обычно неустойчивые соединения, через которые пролегает путь от реагентов к продуктам, называют интермедиатами. Любой интермедиатимеет определенное время жизни, обычно крайне непродолжительное, вплоть до 10 -14 с. На энергетическом профиле реакции ему отвечает минимум (рис. а) (Рис. 1.38. Энегртеческие профили реакции, протекающей через: (а) интермедиат, (б) переходное состояние.). Как правило, интермедиаты могут быть зафиксированы в реакционной смеси спектральными методами, и лишь в редких случаях их удается выделить в индивидуальном виде. Поэтому главную информацию о механизме реакции обычно получают через изучение ее кинетики – определяя константы скорости и рассчитывая параметры активации (энтальпию, энтропию, объем). В этом случае механизм – это модель, которая находится в соответствии с кинетическими данными, модель, которая может быть улучшена, модифицирована, пересмотрена.
В некоторых реакциях интермедиаты не образуются, а переход от реагентов к продуктам протекает последовательно – один из атомов постепенно удаляется, а другой приближается. В таком случае говорят, что реакция протекает через переходное состояние или активированный комплекс. Ему соответствует максимум на энергетическом профиле реакции (Рис. Б).
Дополнение: Лабильные и инертные комплексы
Термодинамическая устойчивость частицы определяется изменением энергии Гиббса для реакции ее диссоциации, либо значением константы устойчивости этого процесса. Кинетическая устойчивость показывает, насколько быстро данная частица вступает во взаимодействие с другими частицами или претерпевает распад. Химическая частица считается инертной , если она вступает в реакцию с периодом полупревращения более 1 минуты. Частицы, реагирующие с более высокой скоростью, называют лабильными . Необходимо помнить, что кинетическая и термодинамическая устойчивость не зависят одна от другой, то есть одно и то же вещество может иметь высокую константу устойчивости и в то же время быть инертным, или, наоборот, лабильным. Некоторые такие примеры приведены в таблице 1.6.
Таблица 1.6. Константы устойчивости и скорости замещения лигандов в циано-комплексах некоторых металлов.
Генри Таубе показал связь кинетической устойчивости октаэдрических комплексов с электронной конфигурацией центрального иона в октаэдрическом поле. Согласно Таубе, лабильными являются комплексы:
· обладающие хотя бы одной вакантной t 2g -орбиталью – они могут ее использовать в реакциях по ассоциативному (A, I a) механизму, либо
· имеющие хотя бы один электрон на e g -орбитали – это способствует реакции по диссоциативному (D, I d) механизму, т.к. удаление электрона с e g -орбитали понижает энергию переходного состояния.
Таким образом, к инертным относятся октаэдрические комплексы хрома(III) (t 2g 3), низкоспиновые комплексы железа(II) (t 2g 6) и железа(III) (t 2g 5), а также комплексы 4d-, 5d-переходных элементов с числом d-электронов больше двух.
КОНЕЦ ДОПОЛНЕНИЯ
Единая классификация неорганических реакций не разработана до сих пор. Условно можно предложить следующую схему (Рис. 1.39. Схема, иллюстрирующая классификацию неорганических реакций):
1) Реакции замещения, присоединения или отщепления лигандов затрагивают изменение координационной сферы металла,
2) Окислительно-восстановительные реакции связаны с изменением электронной конфигурации металла, но не затрагивают его координационное окружение,
3) Реакции координированных лигандов затрагивают изменение лиганда без изменения координационной сферы комплекса.
Реакции замещения. В широком смысле под реакциями замещения понимают процессы замещения одних лигандов в координационной сфере металла другими. Такие реакции могут протекать как с изменением степени окисления, так и без ее изменения. Следуя приведенной выше классификации, мы будем использовать этот термин лишь применительно к реакциям, протекающим без изменения степеней окисления.
Классификация реакций замещения в неорганической химии была разработана Лэнгфордом и Греем. Она основана на определении так называемого предельного механизма, а не на описании конкретного механизма. Первоначально определяют стехиометрический механизм, а затем – внутренний. Стехиометрический механизм – это последовательность элементарных стадий при переходе от исходных веществ к продуктам. Он может быть диссоциативным (D), ассоциативным (A) и обменным (взаимного обмена, I). Диссоциативный и ассоциативный процессы представляют собой как бы два предельных случая, прямо противоположных один другому. Оба процесса протекают в две стадии через образование интермедиата.
Диссоциативный (D)
Процесс двухстадийный, в предельном случае протекает через интермедиат с пониженным КЧ:
ML 6 + L, + Y ¾® ML 5 Y
Ассоциативный (A)
Процесс двухстадийный, характеризуется образованием интермедиата с повышенным КЧ:
ML 6 + Y , ¾® ML 5 Y + L
Взаимного обмена (I)
По этому механизму протекает большинство реакций обмена. Процесс одностадийный и не сопровождается образованием интермедиата. В переходном состоянии реагент и уходящая группа связаны с реакционным центром, входят в его ближайшую координационную сферу, и в процессе реакции происходит вытеснение одной группы другой, обмен двух лигандов:
ML 6 + Y ML 5 Y + L.
Переходное состояние представляет собой либо внешнесферный комплекс, либо, в случае заряженных лигандов, ионную пару MX 5 L + Y - .
Внутренний механизм (a или d ) характеризует процесс замещения лигандов на молекулярном уровне. Он показывает, какой из двух процессов – образование или разрыв связи в переходном состоянии, является лимитирующим. В случае, если скорость реакции определяется образованием связи между реакционным центром и реагентом, говорят об ассоциативной активации. В противном случае, когда лимитирующим фактором служит разрыв связи между реакционным центром и уходящей группой, процесс протекает с диссоциативной активацией. Обращаясь к стехиометрическому механизму, легко заметить, что диссоциативному процессу всегда соответствует диссоциативная активация, ассоциативному – ассоциативная, то есть понятие о внутреннем механизме оказывается информативным лишь в случае механизма взаимного обмена – он может протекать как с диссоциативной (I d), так и с ассоциативной (I a) активацией. В случае механизма взаимного обмена с ассоциативной активацией (I а) скорость реакции зависит от природы Y. В переходном состоянии атом металла прочно связан как с уходящей группой, так и с атакующим нуклеофилом. Примером служит процесс замещения атома хлора на бром и иод в комплексе платины с диэтилентриамином (dien):
Y - ¾¾® + + Cl -
Y = Br, I скорости сильно различаются.
В случае механизма взаимного обмена с диссоциативной активацией (I d) скорость реакции не зависит от природы реагента Y. Атакующая и уходящая группы в переходном состоянии слабо связаны с центральным ионом. По такому механизму протекает замещение воды на амин в аквакомплексах многих переходных металлов, например, никеля:
2+ + Y ¾¾® 2+ + H 2 O
Y = NH 3 , py скорости близки.
Исследование механизмов реакций замещения в комплексах многих металлов находится лишь в первоначальной стадии. Исчерпывающая информация получена лишь для плоско-квадратных комплексов платины и октаэдрических комплексов хрома(III) и кобальта(III). Можно считать твердо установленным, что в комплексах платины(II) замещение протекает по ассоциативному механизму (A, Ia) через интермедиат или переходное состояние в форме тригональной бипирамиды. Октаэдрические комплексы кобальта(III) реагируют диссоциативно (D, I d механизмы). Конкретные примеры таких реакций будут рассмотрены при описании химии этих элементов.
Окислительно-восстановительные реакции. Большинство окислительно-восстановительных процессов представляет собой сложную комбинацию отдельных элементарных стадий, на каждой из которых происходит перенос одного или, значительно реже, двух электронов. Одновременный перенос большего числа электронов в растворах невозможен.
Одноэлектронный перенос может протекать по одному из двух механизмов – внешнесферно, то есть путем туннелирования, либо внутрисферно – через мостиковый лиганд. Внутрисферный механизм реализуется в комплексах, содержащих галогениды, гидроксид-ион, карбоксильные группы, способные выступать в качестве мостиков между металлами. Примером служит реакция между ионами пентамминхлорокобальта(III) и гексааквахрома(II). Процесс можно условно разбить на три стадии – образование гетерометаллического комплекса с мостиковым хлорид-ионом, электронный перенос и распад мостикового комплекса. Образующийся ион 2+ , будучи лабильным, мгновенно превращается в аква-комплекс, а инертный [(H 2 O) 5 CrCl] 2+ не вступает во взаимодействие с водой:

Если в системе нет частиц, которые могли бы выступать в роли мостиковых, процесс протекает внешнесферно:
2+ + 3+ = 3+ + 2+ .
Особо необходимо выделить реакции окислиетльного присоединения и восстановтельного элиминирования, рассмотренные в главе 6.
Реакции координированных лигандов. В данную группу реакций входят процессы модификации лигандов, координированных ионом металла. Так, например, дикетонатные комплексы, подобно свободным дикетонам, можно нитровать, ацилировать, галогенировать. Наиболее интересным и необычным примером реакций координированных лигандов является темплатный синтез – своебразный метод «сборки» лиганда на ионе металла. Примером может служить синтез фталоцианинов из нитрила фталевой кислоты, протекающий в присутствии ионов меди(II), и синтез макроциклического основания Шиффа из 2-аминобензальдегида, протекающий на ионах никеля(II):

В отсутствии металла процесс протекает по другому пути, и в реакционной смеси желаемый продукт присутствует лишь в незначительном количестве. Ион металла выступает в темплатном синтезе в качестве матрицы («темплата»), стабилизирующей один из продуктов, находящихся в равновесии друг с другом, и смещающей равновесие в сторону его образования. Например, в реакции X + Y ¾® образуется смесь продуктов A и B, в которой преобладает В, имеющий более низкую энергию. В присутствии иона металла в продуктах реакции преобладает вещество А в виде комплекса с М (Рис. 1.40. Энергетическая диаграмма взаимодействия X и Y в отсутствии иона металла (слева) и в его присутствии (б)).
Вопросы и задания
1. Какие из перечисленных ниже соединений имеют структуру перовскита? BaTiO 3 , LiNbO 3 , LaCrO 3 , FeTiO 3 , Na 2 WO 4 , CuLa 2 O 4 , La 2 MgRuO 6 . Таблица ионных радиусов приведена в Приложении. Имейте в виду, что в сложных оксидных фазах в позициях В могут располагаться катионы двух различных металлов.
2. Воспользовавшись ТКП, определите, прямыми или обращенными будут следующие шпинели: ZnFe 2 O 4 , CoFe 2 O 4 , Co 3 O 4 , Mn 3 O 4 , CuRh 2 O 4 .
3. Тиоцианат-ион SCN – имеет два донорных центра – жесткий и мягкий. Предположите, какое строение будут иметь тиоцианатные комплексы кальция и меди(I). Почему не удается получить роданид меди(II)?
4. Спектр акваиона Cr 2+ (терм основного состояния 5 D) имеет две полосы (Рис. 1.41. Спектр акваиона Cr 2+), хотя среди термов ближайших возбужденных состояний нет ни одного с такой же мультиплетностью. Чем это объясняется? Какую окраску имеет этот ион?
5. Используя приведенные ниже значения Δο, рассчитайте ЭСКП для следующих комплексов в кДж/моль:
(а) 2– , Δο = 15000 см –1 ,
(б) 2+ , Δο = 13000 см –1 ,
(в) 2– , Δο (для 4–)= 21000 см –1 ,
Энергию спаривания примите равной 19000 см –1 , 1кДж/моль = 83 см –1 . Рассчитайте их магнитные моменты (спиновую составляющую).
6. Используя ТКП, объясните, почему ион CN – реагирует с ионом гексаакважелеза (III) с образованием гексацианоферрата(II), а с ионом гесаакваникеля(II) с образованием тетрацианоникелата(II).
7. Ниже приведены константы реакций последовательного замещения воды в аквакомплексе меди(II) на аммиак: K 1 = 2´10 4 , K 2 = 4´10 3 , K 3 = 1´10 3 , K 4 = 2´10 2 , K 5 = 3´10 –1 , K 6 << 1. Чем объясняется трудность вхождения пятой и шестой молекул аммиака в координационную сферу меди?
8. Как изменяется жесткость катионов при движении по 3d-ряду? Согласуется ли это с порядком изменения констант устойчивости комплексов (ряд Ирвинга-Уильямса, рис..1.34).
9. Объясните, почему ион гексаакважелеза (III) бесцветный, а растворы солей железа (III) окрашены.
10. Предложите механизм реакции 3– + 3– = 4– + 2– , если известно, что введение в раствор роданид-иона приводит к изменению скорости реакции, а от присутствия аммиака скорость практически не зависит. Предложите объяснение этим фактам.
Концепция изменения электронного строения ионов переходных металлов при действии электрического поля окружающих его заряженных частиц была предложена Беккерелем и в дальнейшем развита Х.А. Бете и Дж. Ван Флеком в начале XX в. К описанию электронного строения и свойств комплексных соединений эти представления были применены только в середине XX века Х. Хартманом и модель получила название «теория кристаллического поля» (ТКП).
Основные положения ТКП для комплексов переходных d металлов Рис. 24):
1. - Комплекс существует и устойчив, благодаря электростатическому взаимодействию комплексообразователя с лигандами.
2. - Лиганды рассматриваются без учета их электронного строения в качестве точечных зарядов или диполей.
3. - Под действием электрического поля лигандов валентные пятикратно вырожденные (n -1) d орбитали расщепляются в зависимости от симметрии лигандного окружения.
4. - Распределение валетных электронов металла по расщепленным (n -1) d орбиталям зависит от соотношения энергии спин-спаривания и энергии расщепления.
Рассмотрим, например, изменение энергии пятикратно вырожденных(n -1) d орбиталей центрального иона металла М n + , находящегося в центре координат, под действием октаэдрического поля отрицательно заряженных лигандов [ ML 6 ] z , расположенных на осях координат (Рис. 25). В результате отталкивания валентных электронов металла от отрицательно заряженных лигандов при равномерном распределении отрицательного заряда вокруг металла (сферически симметричное электрическое поле) энергия всех пяти d орбиталей повысится на величину Е 0 по сравнению со свободным М n + ионом. Поскольку d орбитали имеют различную пространственную ориентацию, то при концентрации отрицательных зарядов на лигандах, располо-женных на осях координат, повышение их энергии различается. Повышение энергии d z 2 и d x 2- y 2 орбиталей, направленных к лигандам на осях координат, больше повышения энергии d xy , d xz и d yz орбиталей, направленных между осями координат.
Энергия расщепления пятикратно вырожденных (n -1) орбиталей на двухкратно вырожденные d x 2- y 2, z 2 орбитали и трехкратно вырожденные d xy , xz , yz орбитали называется (Рис. 26) параметром расщепления кристаллическим полем. Поскольку энергия расщепленных d орбиталей в октаэдрическом поле лигандов по сравнению со сферически симметричным электрическим полем не изменяется, то повышение энергии двух d x 2- y 2, z 2 орбиталей происходит на 0.6 D 0 и понижение энергии трех d xy , xz , yz орбиталей на 0.4 D 0 .
Для указания степени вырожденности и симметрии расщепленных под
действием электрического поля лигандов орбиталей металла используют специальные
символы. Трехкратно вырожденные и симметричные относительно центра симметрии и
вращения вокруг осей координат
d xy
,
xz
,
yz
t
2
g
», тогда как двухкратно вырожденные и
также симметричные относительно центра симметрии
d x
2-
y
2,
z
2
орбитали обозначают символом «
e g
». Таким образом, под действие
октаэдрического электрического поля лигандов пятикратно вырожденные (n
-1)
d
орбитали комплексообразователя расщепляются на различные
по энергии трекратно и двухкратно вырожденные
t
2
g
и
e g
орбитали.
Подобное рассмотрение изменения энергии пятикратно вырожденных (n -1) d орбиталей свободного иона металла при тетраэдрическом окружении лигандов в [ ML 4 ] z комплексах показывает (рис. 27) их расщепление также на двукратно (е) и трекратно (t ) вырожденные орбитали, однако, с обратным энергетическим положением. Нижний индекс « g » при обозначении «е» и « t » орбиталей не указавается поскольку тетраэдрический комплекс не имеет центра симметрии. Уменьшение числа лигандов тетраэдрического комплекса по сравнению с октаэдрическим приводит к закономерному уменьшению параметра расщепления кристаллическим полем: D Т = 4/9 D О .
Понижение симметрии лигандного окружения металла, например, тетрагональное искажение октаэдрических [ ML 6 ] z комплексов, связанное с удлинением металл-лиганд связей с аксиальными лигандами [ ML 4 X 2 ] z и образованием в предельном случае плоско-квадратных [ ML 4 ] z комплексов, приводит (рис. 28) к дополни-тельному расщеплению валентных (n -1) d орбиталей металла.
Заполнение валентными электронами расщепленных (n -1) d орбиталей металла происходит в соответствии с принципами Паули и минимума энергии. Для октаэдрических комплексов с d 1 , d 2 и d 3 электронной конфигурацией металла валентные электроны в соответствии с правилом Хунда заселяют t 2 g орбитали с параллльными спинами, приводя к t 2 g 1 , t 2 g 2 и t 2 g 3 электронной структуре комплексов.
Для металлов с d 4 электронной конфигурацией три электрона также заселяют t 2 g орбитали с параллельными спинами. Заселение же четвертого электрона зависит от энергетических затрат на величину энергии спин спаривания (Е сп.-сп.) при заселении t 2 g орбиталей с антипараллельным спином и нарушении правила Хунда, либо преодоления энергии расщепления кристаллическим полем D о при заселении e g орбиталей с параллельным спином в соответствии с правилом Хунда. В первом случае образуется комплекс с t 2 g 4 электронным строением и уменьшенным по сравнению со свободным металлом спиновой мультиплетностью 2 S +1 = 3 (S - сумарный спин), называемых низкоспиновыми . При выполнении правила Хунда и заселении четвертого электрона на e g орбитали образуется комплекс с t 2 g 3 e g 1 электронной структурой и подобной свободному металлу спиновой мультиплет-ностью 2 S +1 = 5. Такие комплексы называют высокоспиновыми.
Аналогично, при распределении валентных d 5 , d 6 и d 7 электронов металлов по t 2 g и e g орбиталям октаждрических комплексов в зависимости от соотношения Е сп.-сп. и D о возможно образование двух типов комплексов:
При Е сп.-сп. > D о обрауются высокоспиновые комплексы с электронной структурой металла t 2 g 3 e g 2 , t 2 g 4 e g 2 , t 2 g 5 e g 2 в соответствии с правилом Хунда и спиновой мультиплетностью, подобной свободному металлу - 2 S +1 = 6, 5, 4;
Е сп.-сп. < D о образуются низкоспиновые комплексы с электронной структурой металла t 2 g 5 e g 0 , t 2 g 6 e g 0 , t 2 g 6 e g 1 и пониженной по сравнению со свободным металлом спиновой мультиплетностью 2 S +1 = 2, 1, 2.
Комплексы металлов с d 8 , d 9 и d 10 электронной конфигурацией характеризуются одним типом распределения электронов - t 2 g 6 e g 2 , t 2 g 6 e g 3 , t 2 g 6 e g 4 со спиновой мультиплетностью, подобной свободному металлу: 2 S +1 = 3, 2 и 0.
Таким образом, параметр D , характеризующий расщепление (n -1) d орбиталей металла под действием электрического поля лигандов, является одной из основных характеристик изменения свойств комплексов по сравнению со свободным ионом металла. Именно величина параметра D определяет для ряда электронных конфигураций металла определяет возможность образования высоко- или низкоспиновых комплексов с различным распределением электронов по расщепленным орбиталям и различными свойствами.
Величина параметра расщепления кристаллическим полем D зависит от природы металла комплексообразователя, окружающих его лигандов и их пространственного положения вокруг комплексообразователя:
1. Лиганды в порядке увеличения параметра D для комплексов одного металла и подобного геометрического строения распологаются в так называемом спектро-химическом ряду: I - < Br - < Cl - < F - < OH - < C 2 O 4 2- ~ H 2 O < NCS - < NH 3 ~ En < NO 2 - < CN - < CO . В начале ряда расположены лиганды «слабого поля» - галогенид ионы, гидроксид и оксалат ионы, вода, образующие преимущественно высокоспиновые комплексы. Лиганды, в правой части ряда: окись углерода, цианид и нитрит ионы называются лигандами «сильного поля» и для них типично образование низкоспиновых комплексов. Для лигандов середины ряда - роданид иона, аммиака, этилендиамина в зависимости от природы металла образуются высоко- или низкоспиновые комплексы.
2. Увеличение эффективности действия электрического поля лигандов на d орбитали металла с увеличением их размера в ряду 3 d << 4 d < 5 d , а также увеличения степени окисления металла приводит к увеличению параметра D в ряду: Mn (II ) < Ni (II ) < Co (II ) < Fe (II ) < V (II ) < Fe (III ) < Co (III ) < Mn (IV ) < Mo (III ) < Rh (III ) < Ru (III ) < Pd (IV ) < Ir (III ) < Pt (IV ).
3. Параметр D для тетраэдрических комплексов составляет только 4/9 от параметра D октаэдрических комплексов.
Комплексы «тяжелых» 4 d и 5 d металлов практически не зависимо от природы лигандов образуют преимущественно низкоспиновые комплексы, тогда как образование низко- или выскоспиновых комплексов «легких» 3 d металлов в основном определяется силой поля лигандов.
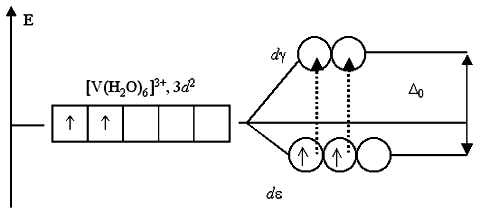
В отличие от МВС, теория кристаллического поля для обоснования различия магнитных свойств комплексов одного и того же иона металла с различным лигандным окружением, например, диамагнитного [ Fe (CN ) 6 ] 4- и парамагнитного [ Fe (H 2 O ) 6 ] 2+ не использует гипотезу о их внутриорбитальном (d 2 sp 3 гибридизация) и требующем больших энергетических затрат внешнеорбитальном (sp 3 d 2 гибриди-зация) строении. Различие в магнитных свойствах определяется низко- и высокоспиновым характером распределения 6-ти валетных электронов Fe (II ) по расщепленным t 2 g и e g орбиталям (рис. 29). Являясь лигандами сильного и слабого поля, цианид-ионы и молекулы воды образуют с Fe (II ) низко- и высокоспиновыекомплексы с t 2 g 6 e g 0 и t 2 g 4 e g 2 распределением электронов, что и определяет диамагнетизм [ Fe (CN ) 6 ] 4- и парамагнетизм [ Fe (H 2 O ) 6 ] 2+ комплексов.
Расщепление пятикратно вырожденных (n
-1)
d
орбиталей
металла в комплексах и изменение параметра
D
в зависимости от природы лигандов
определяет характерную окраску комплексов как в твердом состоянии, так и в
растворах. При поглощении комплексом электромагнитного излучения в видимой
области спектра (400-750) нм, энергия квантов которого Е
равна
величине
D
, происходит перенос электрона с
t
2
g
на
e g
орбитали.
Не поглощенное электромагнитное излучение видимой области спектра и определяет
окраску комплекса в соответствии с «цветовым кругом Ньютона» (Рис. 30), показывающим
основной и дополнительный цвет видимого излучения.
Аквакомплекс титана(III ) [ Ti (H 2 O ) 6 ] 3+ c t 2 g 1 e g 0 электронным распределением в результате фотовозбуждения, соответствующего переходу электрона на более высокоэнергетические e g орбитали:
3+ (t 2g 1 e g 0) + h n = * 3+ (t 2g 0 e g 1)
поглощает кванты света в желтой области спектра, что приводит к его фиолетовой окраске. Изменение лигандного окружения иона металла в соответствии с положением лиганда в спектрохимическом ряду приводит к изменению параметра D и, как следствие этого, к изменению энергии и длины волны поглощаемых комплексом квантов и к характеристической окраске комплекса - например, в ряду [ CuCl 4 ] 2- , [ Cu (H 2 O ) 4 ] 2+ , [ Cu (NH 3 ) 4 ] 2+ цвет комплексов изменяется от зеленого к голубому и фиолетовому.
Наряду с энергией расщепления кристаллического поля D , важную роль в ТКП играет также энергия стабилиции кристаллическим полем (ЭСКП) - выйгрыш в энергии при распределении электронов по расщепленным в комплексе (n -1) d орбиталям металла по сравнению с энергией пятикратно вырожденных (n -1) d орбиталей металла в эквивалентном сферическом электрическом поле (Рис. 31, 32).
ЭСКП октадрических и тетраэдрических комплексов.|
M n+ |
Октаэдрические комлексы |
Тетраэдрические комплексы |
|
|
Низкоспиновые |
Высокоспиновые |
Высокоспиновые |
|
|
0.4 D o |
0.6 D т |
||
|
0.8 D o |
1.2 D т |
||
|
1.2 D o |
0.8 D т |
||
|
d 4 |
1.6 D o |
0.6 D o |
0.4 D т |
|
d 5 |
2.0 D o |
0 D o |
0 D т |
|
d 6 |
2.4 D o |
0.4 D o |
0.6 D т |
|
d 7 |
1.8 D o |
0.8 D o |
1.2 D т |
|
d 8 |
1.2 D o |
0.8 D т |
|
|
d 9 |
0.6 D o |
0.4 D т |
|
|
d 10 |
0 D o |
||
Оценку величины ЭКСП комплекса получают на основании диаграмм
расщепления (n
-1)
d
орбиталей металла в электрическом поле лигандов,
показывающих уменьшение или повышение энергии системы по сравнению со
сферическим электрическим полем при заселении электронами расщепленных (n
-1)
d
орбиталей. Для октаэдри-ческих [
ML
6
]
z
комплексов (Рис. 32) заселение каждым электроном
t
2
g
орбиталей приводит к выйгрышу энергии системы на 0.4
D
о
, заселение
же
e g
требует затрат энергии 0.6
D
о
. Для тетраэдрических [
ML
4
]
z
комплексов с противоположным энергетическим положением
e
и
t
орбиталей металла заселение каждым электроном
расщепленных
e
и
t
орбиталей сопровождается понижением и повышением энергии системы на 0.6
D
т
и 0.4
D
т
.
Являясь отражением термодинамической устойчивости комплеков, оценки их величины ЭСКП согласуются с экспериментальными данными изменения энергии кристаллической решетки для высокоспиновых гексафторидных комплексов 3 d металлов (Рис. 33).
Величины ЭСКП позволяют установить наиболее предпочтительный координационный изомер (Рис. 34), например [ Cu (NH 3 ) 6 ][ NiCl 4 ] или [ Ni (NH 3 ) 6 ][ CuCl 4 ]. Для этого рассчитывают разницу ЭСКП для комплексного катиона и аниона изомеров. Величина ЭСКП [ Cu (NH 3 ) 6 ] 2+ и [ NiCl 4 ] 2- составляет 0.6 D о и 0.8 D т соответственно. Учитывая, что D т = 4/9 D o , разница между величинами ЭСКП [ Cu (NH 3 ) 6 ] 2+ и [ NiCl 4 ] 2- будет составлять 19/45 D o . Аналогично, величины ЭСКП [ Ni (NH 3 ) 6 ] 2+ и [ CuCl 4 ] 2- составляет 1.2 D о и 0.4 D т , а разница между ними 28/45 D o . Большая разница ЭСКП комплексных катиона [ Ni (NH 3 ) 6 ] 2+ и аниона [ CuCl 4 ] 2- по сравнению с [ Cu (NH 3 ) 6 ] 2+ и [ NiCl 4 ] 2- показывает более предпочтительное образование изомера состава [ Ni (NH 3 ) 6 ][ CuCl 4 ].
Наряду с магнитными и оптическими свойствами, влияния электронного строения металла на термодинамическую устойчивость комплексов, ТКП предсказывает искажение геометрического строения комплексов при не равномерном распределении электронов по расщепленным (n -1) d орбиталям металла (Рис. 35). В отличие от правильного октаэдрического строения [ Co (CN ) 6 ] 3- с t 2 g 6 e g 0 электронным распределением, тетрагональное искажение аналогичного комплекса [ Cu (CN ) 6 ] 4- с t 2 g 6 e g 3 электронным распределением, содержащего 3 электрона на 2-х кратно вырожденных e g орбиталях, приводит к эффективной трансформации октаэдрического в плоско-квадратный комплекс:
4- = 2- + 2CN - .
Все выше сказанное показывает, что относительная простота и широкие возможности ТКП для объяснения и прогнозирования физико-химических свойств комплексов определяют большую популярность это модели описания химической связи в комплесных соединениях. В тоже время, акцентируя внимание на изменении электронной структуры металла при комплексообразовании, ТКП не учитывает электронное строение лигандов, рассматривая их в качестве точечных отрицательных зарядов или диполей. Это приводит к ряду ограничений ТКП при описании электронного строения комплексов. Например, в рамках ТКП трудно объяснить положение ряда лигандов и металлов в спектрохимических рядах, что связано с определенной степенью ковалентности и возможность образования кратных металл-лиганд связей. Эти ограничения устраняются при рассмотрении электронного строения комплексных соединений более сложным и менее наглядным методом молекулярных орбиталей.
Теория валентных связей была первой из квантовомеханических теорий, использованной для приближенного объяснения характера химических связей в комплексных соединениях. В основе ее применения лежала идея о донорно-акцепторном механизме образования ковалентных связей между лигандом и комплексообразователем. Лиганд считается донорной частицей , способной передать пару электронов акцептору – комплексообразователю , предоставляющему для образования связи свободные квантовые ячейки (атомные орбитали) своих энергетических уровней.
Для образования ковалентных связей между комплексообразователем и лигандами необходимо, чтобы вакантные s -, p - или d -атомные орбитали комплексообразователя подверглись гибридизации определенного типа. Гибридные орбитали занимают в пространстве определенное положение, причем их число соответствует координационному числу комплексообразователя.
При этом часто происходит объединение неспаренных электронов комплексообразователя в пары, что позволяет высвободить некоторое число квантовых ячеек – атомных орбиталей, которые затем участвуют в гибридизации и образовании химических связей.
Неподеленные пары электронов лигандов взаимодействуют с гибридными орбиталями комплексообразователя, и происходит перекрывание соответствующих орбиталей комплексообразователя и лиганда с появлением в межъядерном пространстве повышенной электронной плотности. Электронные пары комплексообразователя, в свою очередь, взаимодействуют с вакантными атомными орбиталями лиганда, упрочняя связь по дативному механизму . Таким образом, химическая связь в комплексных соединениях является обычной ковалентной связью, достаточной прочной и энергетически выгодной .
Электронные пары, находящиеся на гибридных орбиталях комплексообразователя, стремятся занять в пространстве такое положение, при котором их взаимное отталкивание будет минимально. Это приводит к тому, что структура комплексных ионов и молекул оказывается в определенной зависимости от типа гибридизации .
Рассмотрим образование некоторых комплексов с позиций теории валентных связей. Прежде всего отметим, что валентные орбитали атомов комплексообразователей близки по энергии:
E (n - 1)d » E ns » E np » E nd
|
Тип гибридизации |
Геометрия комплекса |
||
|
линейная |
-
|
||
|
треугольная |
- |
||
|
тетраэдр |
2-
|
||
|
2-
|
|||
|
sp 3 d (z 2) |
тригональная бипирамида |
||
|
sp 3 d (x 2 - y 2) |
квадратная пирамида |
3-
|
|
|
sp
3 d
2 , |
3+ |
||
|
sp 3 d 3 |
пентагональная бипирамида |
4-
|
Например, катион 2+ включает комплексообразователь цинк(II). Электронная оболочка этого условного иона имеет формулу 3d 10 4s 0 4p 0 и может быть условно изображена так:
Вакантные 4s
- и 4p
-орбитали атома цинка(II) образуют четыре sp
3 -гибридные орбитали, ориентированные к вершинам тетраэдра.
Каждая молекула аммиака имеет неподеленную пару электронов у атома азота. Орбитали атомов азота, содержащие неподеленные пары электронов, перекрываются с sp
3 -гибридными орбиталями цинка(II), образуя тетраэдрический комплексный катион тетраамминцинка(II) 2+ : 
Поскольку в ионе 2+ нет неспаренных электронов, то он проявляет диамагнитные свойства.
Тетрахлороманганат(II)-ион 2-
содержит пять неспаренных электронов на 3d
-орбитали и вакантные 4s
- и 4p
-орбитали. Вакантные орбитали образуют sp
3 -гибридные орбитали, которые перекрываются с p
-атомными орбиталями хлорид-ионов:
Полученный таким образом тетраэдрический ион 2- является парамагнитным , так как содержит пять неспаренных электронов.
Применяя обычный алгоритм предсказания типа гибридизации атомных орбиталей в рамках метода валентных связей, можно определить геометрию комплексов разного состава. Для этого прежде всего необходимо написать электронную формулу валентного уровня и построить схему распределения электронов по квантовым ячейкам. Например, для нейтрального атома никеля:

Переход 4s
-электронов на 3d
-подуровень превращает парамагнитный
атом Ni 0 в диамагнитную
частицу Ni*:

Полученные вакантные орбитали подвергаются гибридизации, образуя тетраэдрическую конфигурацию. Так построен тетраэдрический диамагнитный комплекс тетракарбонилникель (КЧ = 4), который характеризуется значительной устойчивостью.
Если комплексообразователем служит никель(II) с электронной конфигурацией 3d
8 4s
0 4p
0 , то надобность в перемещении электронов с 4s
-подуровня перед гибридизацией отпадает, так как для реализации координационного числа 4 имеется достаточное число вакантных орбиталей:

Такое строение имеет неустойчивый парамагнитный
комплекс тетрабромоникколат(II)-ион 2-
. Однако при объединении двух электронов 3d
-подуровня в пару и превращении одной из квантовых ячеек этого подуровня в вакантную меняется и тип гибридизации, и характеристика получаемого комплекса:

Тип гибридизации dsp
2 и плоскоквадратная форма комплекса реализуются при образовании устойчивого диамагнитного
комплекса тетрацианоникколат(II)-иона 2-
(КЧ = 4):
Если синтез цианидного комплекса вести в условиях избытка лиганда, можно реализовать координационное число 5:
Устойчивый диамагнитный
комплекс пентацианоникколат(II)-ион 3-
имеет форму квадратной пирамиды:

Октаэдрический комплекс никеля(II) 2+ , хотя и парамагнитен
, но достаточно устойчив. Его образование обусловлено sp
3 d
2 -гибридизацией атомных орбиталей никеля:

Если в гибридизации участвуют атомные орбитали внешнего d
-подуровня, комплекс, как правило, в значительной степени парамагнитен
и называется внешнеорбитальным
или высокоспиновым
. Строение таких комплексов может отвечать типу гибридизации, например, sp
3 d
2 .
Такие комплексы, при образовании которых имеет место гибридизация с участием атомных орбиталей предвнешнего d
-подуровня, называются внутриорбитальными
или низкоспиновыми
и, как правило диамагнитны
или слабо парамагнитны
(все или почти все электроны комплексообразователя спарены, а тип гибридизации, например, d
2 sp
3 или dsp
2).
При рассмотрении комплексов железа(II) обнаруживаются и внешнеорбитальные, и внутриорбитальные комплексы.


Приведенная схема показывает, как образуются парамагнитный высокоспиновый гексафтороферрат(II)-ион 4- и диамагнитный низкоспиновый гексацианоферрат(II)-ион 4- .
Сама по себе теория валентных связей не дает ответа на вопрос, какой вид комплекса образуется в каждом конкретном случае, так как этот метод не учитывает влияния природы лиганда. Поэтому метод валентных связей должен обязательно дополняться данными о магнитных свойствах комплекса либо сведениями о влиянии лиганда на характер образующегося комплекса.
.Теория кристаллического поля пришла на смену теории валентных связей в 40-х годах XX столетия. В чистом виде она сейчас не применяется, так как не может объяснить образование ковалентных связей в комплексных соединениях и совершенно не учитывает истинного состояния лигандов (например, их действительных размеров) даже в случае взаимодействий, близких к чисто электростатическим.
Уже с середины 50-х годов упрощенная теория кристаллического поля была заменена усовершенствованной теорией поля лигандов , учитывающей ковалентный характер химических связей между комплексообразователем и лигандом.
Однако наиболее общий подход к объяснению образования комплексных соединений дает теория молекулярных орбиталей (МО), которая в настоящее время превалирует над всеми остальными. Метод молекулярных орбиталей предусматривает и чисто электростатическое взаимодействие при отсутствии перекрывания атомных орбиталей, и всю совокупность промежуточных степеней перекрывания.
Рассмотрим основные понятия теории кристаллического поля , которая, как и теория валентных связей, все еще сохраняет свое значение для качественного описания химических связей в комплексных соединениях из-за большой простоты и наглядности.
В теории кристаллического поля химическая связь комплексообразователь – лиганд считается электростатической . В соответствии с этой теорией лиганды располагаются вокруг комплексообразователя в вершинах правильных многогранников (полиэдров ) в виде точечных зарядов . Реальный объем лиганда теорией во внимание не принимается.
Лиганды, как точечные заряды, создают вокруг комплексообразователя электростатическое поле (“кристаллическое поле”, если рассматривать кристалл комплексного соединения, или поле лигандов ), в котором энергетические уровни комплексообразователя и прежде всего d -подуровни расщепляются , и их энергия изменяется. Характер расщепления, энергия новых энергетических уровней зависит от симметрии расположения лигандов (октаэдрическое, тетраэдрическое или иное кристаллическое поле). Когда в качестве лигандов координируются молекулы H 2 O, NH 3 , CO и другие, их рассматривают как диполи , ориентированные отрицательным зарядом к комплексообразователю.
Рассмотрим случай октаэдрического расположения лигандов (например, 3- или 3+). В центре октаэдра находится атом-комплексообразователь М(+n ) с электронами на d -атомных орбиталях, а в его вершинах – лиганды в виде точечных отрицательных зарядов (например, ионы F - или полярные молекулы типа NH 3). В условном ионе М(+n ), не связанном с лигандами, энергии всех пяти d -АО одинаковы (т.е. атомные орбитали вырожденные ).
Однако в октаэдрическом поле лигандов d
-АО комплексообразователя попадают в неравноценное
положение. Атомные орбитали d
(z
2) и d
(x
2 -
y
2), вытянутые вдоль осей координат, ближе всего подходят к лигандам. Между этими орбиталями и лигандами, находящимися в вершинах октаэдра, возникают значительные силы отталкивания
, приводящие к увеличению энергии орбиталей. Иначе говоря, данные атомные орбитали подвергаются максимальному воздействию поля лигандов
. Физической моделью такого взаимодействия может служить сильно сжатая пружина.

Другие три d
-АО – d
(xy
), d
(xz
) и d
(yz
), расположенные между осями координат и между лигандами, находятся на более значительном расстоянии от них. Взаимодействие таких d
-АО с лигандами минимально, а следовательно – энергия d
(xy
), d
(xz
) и d
(yz
)-АО понижается по сравнению с исходной.

Таким образом, пятикратно вырожденные d
-АО комплексообразователя, попадая в октаэдрическое поле лигандов
, подвергаются расщеплению
на две группы новых орбиталей – трехкратно вырожденные орбитали
с более низкой энергией, d
(xy
), d
(xz
) и d
(yz
), и двукратно вырожденные орбитали
с более высокой энергией, d
(z
2) и d
(x
2 -
y
2). Эти новые группы d
-орбиталей с более низкой
и более высокой энергией
обозначают d
e
и d
g
:
Разность энергий двух новых подуровней d e и d g получила название параметра расщепления D 0:
E 2 – E 1 = D 0
Расположение двух новых энергетических подуровней d e и d g по отношению к исходному (d -АО) на энергетической диаграмме несимметричное :
(Е 2 – Е 0) > (Е 0 – Е 1).
Квантово-механическая теория
требует, чтобы при полном заселении новых энергетических уровней электронами общая энергия осталась без изменения
, т.е. она должна остаться равной
Е
0 .
Иначе говоря, должно выполняться равенство
4(Е 2 – Е 0) = 6(Е 0 – Е 1),
где 4 и 6 – максимальное число электронов на d g - и d e -АО. Из этого равенства следует, что
(Е
2 – Е
0) / (Е
0 – Е
1) = 3/2 и
(Е
2 – Е
1) / (Е
0 – Е
1 >) = 5/2, или
D
0 / (Е
0 – Е
1) = 5/2, откуда (Е
0 – Е
1) = 2/5 ´
D
0 >.
Размещение каждого электрона из шести максимально возможных на d
e
-орбитали вызывает уменьшение
(выигрыш
) энергии
на 2/5 D
0 . Наоборот, размещение каждого электрона из четырех возможных на d
g
-орбитали вызывает увеличение
(затрату
) энергии
на 3/5 D
0 . Если заселить электронами d
e
- и d
g
-орбитали полностью, то никакого выигрыша
энергии не будет
(как не будет и дополнительной затраты энергии
): 4 ´
3/5 ´
D
0 -
6 ´
2/5 ´
D
0 = 0. Но если исходная d
-АО заселена только частично
и содержит от 1 до 6 электронов, и эти электроны размещаются только на d
e
-АО, то мы получим значительный выигрыш энергии
. Специфика каждого из лигандов сказывается в том, какое поле данный лиганд создает – сильное
или слабое
. Чем сильнее поле
лигандов, чем больше
значение параметра расщепления
D
0 . Изучение параметра расщепления, как правило, основано на спектроскопических
исследованиях. Длины волн полос поглощения
комплексов l
в кристаллическом состоянии или в растворе, обусловленные переходом электронов с d
e
- на d
g
-АО, связаны с параметром расщепления
D
0 следующим образом: n
= 1 / l
;
D
где постоянная Планка h
равна 6,626 ´
10 -
34 Дж. с; Параметр расщепления
, помимо типа лиганда, зависит от степени окисления
и природы
комплексообразователя. При увеличении заряда ядра
атома-комплексообразователя D
0 тоже растет. Катионы гексаамминкобальта(III) 3+ , гексаамминродия(III) 3+ , гексаамминиридия(III) 3+ (Z
= 27, 45 и 77) характеризуются параметрами расщепления, равными 22900, 34100 и 41000 см -1 .
Зависимость D
0 от природы лигандов более разнообразна. В результате исследования многочисленных комплексных соединений было установлено, что по способности увеличивать параметр расщепления металлов-комплексообразователей, находящихся в своих обычных степенях окисления, наиболее распространенные лиганды можно расположить в следующий спектрохимический ряд
, вдоль которого значение D
0 монотонно растет: Таким образом, наиболее сильное электростатическое поле вокруг комплексообразователя и самое сильное расщепление d
-АО вызывают лиганды NO 2 - , CN -
и CO.
Рассмотрим распределение электронов по d
e
- и d
g
-орбиталям в октаэдрическом поле лигандов. Заселение d
e
- и d
g
-орбиталей происходит в полном соответствии с правилом Гунда
и принципом Паули
. При этом независимо от значения параметра расщепления первые три электрона занимают квантовые ячейки d
e
-подуровня: Если число электронов на d
-подуровне комплексообразователя больше трех, для размещения их по расщепленным подуровням появляется две возможности. При низком значении параметра расщепления (слабое поле лигандов) электроны преодолевают энергетический барьер, разделяющий d
e
- и d
g
-орбитали; четвертый, а затем и пятый электроны заселяют квантовые ячейки d
g
-подуровня. При сильном поле лигандов и высоком значении D
0 заселение четвертым и пятым электроном d
g
-подуровня исключено; происходит заполнение d
e
-орбиталей. При слабом поле лигандов
заселяющие квантовые ячейки 4 или 5 электронов имеют параллельные спины
, поэтому получаемый комплекс оказывается сильно парамагнитен
. В сильном поле лигандов
образуются одна, а затем две электронные пары на d
e
-подуровне, так что парамагнетизм
комплекса оказывается гораздо слабее. Шестой, седьмой и восьмой электроны в случае слабого поля оказываются снова на d
e
-подуровне, дополняя конфигурации до электронных пар (одной в случае d
6 , двух – d
7 и трех – d
8): В случае сильного поля лигандов шестой электрон заселяет d
e
-АО, приводя к диамагнетизму
комплекса, после чего седьмой и восьмой электроны поступают на d
g
-подуровень: Очевидно, при восьмиэлектронной конфигурации различия в строении
между комплексами с лигандами слабого
и сильного поля исчезают
. Заселение орбиталей девятым и десятым электроном также не различается для комплексов обоих типов: Вернемся к рассмотрению электронного строения октаэдрических комплексных ионов 3+ и 3-
. В соответствии с расположением в спектрохимическом ряду
, аммиак NH 3 относится к числу лигандов сильного поля
, а фторид-ион F -
– слабого поля
. Следовательно, заселение электронами атомных орбиталей в данных комплексах будет происходит по схеме: В анионе 3-
лиганды F -
создают слабое кристаллическое поле (D
0 = 13000 см -
1), и все электроны исходной 3d
6 -АО размещаются на d
e
- и d
g
-орбиталях без какого-либо спаривания. Комплексный ион является высокоспиновым
и содержит четыре неспаренных электрона, поэтому он парамагнитен
. В ионе 3+ лиганды NH 3 создают сильное кристаллическое поле (D
0 = 22900 см -
1), все 3d
6 -электроны размещаются на более энергетически выгодной d
e
-орбитали. Переход электронов с d
e
- на d
g
-орбитали невозможен
из-за слишком высокого энергетического барьера
. Поэтому данный комплексный катион является низкоспиновым
, он не содержит неспаренных электронов и диамагнитен
. Аналогичным образом могут быть представлены схемы распределения электронов по орбиталям в октаэдрическом поле для ионов 2+ и 4-
: Лиганды H 2 O создают слабое поле; обмен электронами между d
e
- и d
g
-орбиталями не вызывает затруднений и поэтому число неспаренных электронов в комплексном ионе такое же, как и в условном ионе Fe +II . Получаемый аквакомплекс – высокоспиновый, парамагнитный
. Многие комплексные соединения в кристаллическом состоянии и водном растворе отличаются яркой окраской. Так, водный раствор, содержащий катионы 2+ , окрашен в интенсивно синий цвет, катионы 3+ придают раствору фиолетовую окраску, а катионы 2+ -
красную. Теория кристаллического поля позволяет объяснить появление той или иной окраски у комплексных соединений. Если через раствор или кристаллический образец вещества пропускать свет видимой части спектра
, то в принципе возможны три варианта физического поведения образца: отсутствие поглощения света
любой длины волны (образец вещества бесцветен
, хотя может иметь полосы поглощения в ультрафиолетовой области спектра); полное поглощение света
во всем интервале длин волн (образец будет казаться черным
); наконец, поглощение света
только определенной длины волны
(тогда образец будет иметь цвет, дополнительный к поглощенному
узкому участку спектра). Таким образом, цвет раствора или кристаллов определяется частотой полос поглощения
видимого света: Поглощение квантов света комплексами (например, имеющими октаэдрическое строение) объясняется взаимодействием света с электронами, находящимися на d
e
-подуровне, сопровождаемое их переходом на вакантные орбитали d
g
-подуровня. Например, при пропускании света через водный раствор, содержащий катионы гексаакватитана(III) 3+ , обнаруживается полоса поглощения света в желто-зеленой области спектра (20300 см -
1 , l
»
500 нм). Это связано с переходом единственного электрона комплексообразователя с d
e
-АО на d
g
-подуровень: Поэтому раствор, содержащий 3+ , приобретает фиолетовый цвет (дополнительный к поглощенному желто-зеленому). Раствор соли ванадия Cl 3 имеет зеленый цвет. Это также обусловлено соответствующими переходами электронов при поглощении ими части энергии светового луча. В основном состоянии, при электронной конфигурации ванадия(III) 3d
2 , два неспаренных электрона занимают d
e
-подуровень: Существует всего два варианта перехода двух электронов
на d
g
-подуровень: либо оба
электрона занимают d
g
-АО, либо только один
из них. Любые другие переходы электронов, связанные с уменьшением суммарного спина, запрещены. Если комплексообразователь имеет электронную конфигурацию d
0 или d
10 , то переходы электронов
с d
e
- на d
g
-подуровень или наоборот невозможны
либо из-за отсутствия электронов
, либо из-за отсутствия вакантных орбиталей
. Поэтому растворы комплексов с такими комплексообразователями, как Sc(III), Cu(I), Zn(II), Cd(II) и т.п., не поглощают энергии в видимой части спектра и кажутся бесцветными
: Избирательность поглощения света зависит не только от комплексообразователя
и степени его окисления
, но и от вида лигандов
. При замене в комплексном соединении лигандов, находящихся в левой части спектрохимического ряда, на лиганды, создающие сильное
электростатическое поле, наблюдается увеличение
доли энергии, поглощаемой электронами из проходящего света и как следствие – уменьшение
длины волны соответствующей полосы поглощения. Так, водный раствор, содержащий катионы тетрааквамеди(II) 2+ , окрашен в голубой цвет, а раствор сульфата тетраамминмеди(II) 2+ имеет интенсивно синюю окраску. ________________________ Повторить:
>>>
Приложения
Выигрыш энергии за счет преимущественного заселения
электронами d
e
-атомных орбиталей называют энергией стабилизации комплекса полем лигандов
.
скорость света с
= 3 ´
10 10 см/с.
Единица измерения
D
0 – та же, что у волнового числа n
: см -
1 , что приближенно отвечает 12 Дж/моль.
В комплексных соединениях, включающих комплексообразователи одного и того же периода и в одинаковой степени окисления, с одними и теми же лигандами, параметр расщепления примерно одинаков. С ростом степени окисления комплексообразователя значение D
0 увеличивается
. Так, для аквакомплексов 2+ и 2+ значение параметра расщепления составляет 7800 и 10400 см -
1 , а для 3+ и 3+ -
13700 и 21000 см -
1 соответственно.
I -
Br -
Cl -
»
NCS -
NO 3 -
F -
OH -
H 2 O »
H -
NH 3 NO 2 -
CN -
»
NO »
CO.









Наоборот, лиганды CN -
вызывают значительное расщепление d
-АО, составляющее 33000 см -
1 . Это значит, что существует сильная тенденция к размещению всех электронов
на d
e
-орбиталях. Выигрыш энергии
, получаемый при таком заселении орбиталей, много больше энергетических затрат, обусловленных спариванием электронов.

Указанным переходам электронов, получивших избыточную энергию, соответствует полоса поглощения
около 400 нм в спектре поглощения раствора хлорида гексаакваванадия(III). Поглощение пурпурно-фиолетовой области спектра дает дополнительный цвет раствора – ярко-зеленый
.

И Джоном Ван Флеком для описания низших состояний катионов переходных металлов, окруженных лигандами - как анионами, так и нейтральными молекулами. Теория кристаллического поля была в дальнейшем объединена [и усовершенствована] с теорией (делокализованных) молекулярных орбиталей в более общую, учитывающую частичную ковалентность связи металл-лиганд в координационных соединениях .
Теория кристаллического поля позволяет предсказать или интерпретировать оптические спектры поглощения и спектры электронного парамагнитного резонанса кристаллов и комплексных соединений, а также энтальпий гидратации и устойчивости в растворах комплексов переходных металлов.
Обзор теории кристаллического поля [ | ]
Согласно ТКП, взаимодействие между переходным металлом и лигандами возникает вследствие притяжения между положительно заряженным катионом металла и отрицательным зарядом электронов на несвязывающих орбиталях лиганда. Теория рассматривает изменение энергии пяти вырожденных d -орбиталей в окружении точечных зарядов лигандов. По мере приближения лиганда к иону металла, электроны лиганда становятся ближе к некоторым d -орбиталям, чем к другим, вызывая потерю вырожденности. Электроны d -орбиталей и лигандов отталкиваются друг от друга как заряды с одинаковым знаком. Таким образом, энергия тех d -электронов, которые ближе к лигандам, становится выше, чем тех, которые дальше, что приводит к расщеплению уровней энергии d -орбиталей.
На расщепление влияют следующие факторы:
- Природа иона металла.
- Степень окисления металла. Чем выше степень окисления, тем выше энергия расщепления.
- Расположение лигандов вокруг иона металла.
- Природа лигандов, окружающих ион металла. Чем сильнее эффект от лигандов, тем больше разность между высоким и низким уровнем энергии.
Самый распространённый вид координации лигандов - октаэдрическая , при которой шесть лигандов создают кристаллическое поле октаэдрической симметрии вокруг иона металла. При октаэдрическом окружении иона металла с одним электроном на внешней оболочке d-орбитали разделяются на две группы с разностью энергетических уровней Δ окт (энергия расщепления ), при этом энергия у орбиталей d xy , d xz и d yz будет ниже, чем у d z 2 и d x 2 -y 2 , так как орбитали первой группы находятся дальше от лигандов и испытывают меньшее отталкивание. Три орбитали с низкой энергией обозначаются как t 2g , а две с высокой - как e g .
Следующими по распространённости являются тетраэдрические комплексы, в которых четыре лиганда образуют тетраэдр вокруг иона металла. В этом случае d -орбитали также разделяются на две группы с разностью энергетических уровней Δ тетр. В отличие от октаэдрической координации, низкой энергией будут обладать орбитали d z 2 и d x 2 -y 2 , а высокой - d xy , d xz и d yz . Кроме того, так как электроны лигандов не находятся непосредственно в направлении d -орбиталей, энергия расщепления будет ниже, чем при октаэдрической координации. С помощью ТКП также можно описать плоскоквадратную и другие геометрии комплексов.
Разность энергетических уровней Δ между двумя или более группами орбиталей зависит также от природы лигандов. Некоторые лиганды вызывают меньшее расщепление, чем другие, причины чего объясняет. Спектрохимический ряд - полученный опытным путём список лигандов, упорядоченных в порядке возрастания Δ:
Степень окисления металла также влияет на Δ. Металл с более высокой степенью окисления ближе притягивает лиганды за счёт большей разности зарядов. Лиганды, находящиеся ближе к иону металла, вызывают большее расщепление.
Низко- и высокоспиновые комплексы [ | ]
Лиганды, вызывающие большое расщепление d -уровней, например CN − и CO, называются лигандами сильного поля . В комплексах с такими лигандами электронам невыгодно занимать орбитали с высокой энергией. Следовательно, орбитали с низкой энергией полностью заполняются до того, как начинается заполнение орбиталей с высокой энергией. Такие комплексы называются низкоспиновыми . Например, NO 2 − - лиганд сильного поля, создающий большое расщепление. Все 5 d -электронов октаэдрического иона 3− будут располагаться на нижнем уровне t 2g .
Напротив, лиганды, вызывающие малое расщепление, например I − и Br − , называются лигандами слабого поля . В этом случае легче поместить электроны в орбитали с высокой энергией, чем расположить два электрона в одной орбитали с низкой энергией, потому что два электрона в одной орбитали отталкивают друг друга, и затраты энергии на размещение второго электрона в орбитали выше, чем Δ. Таким образом, прежде чем появятся парные электроны, в каждую из пяти d -орбиталей должно быть помещёно по одному электрону в соответствии с правилом Хунда . Такие комплексы называются высокоспиновыми . Например, Br − - лиганд слабого поля, вызывающий малое расщепление. Все 5 d -орбиталей иона 3− , у которого тоже 5 d -электронов, будут заняты одним электроном.
Энергия расщепления для тетраэдрических комплексов Δ тетр примерно равна 4/9Δ окт (для одинаковых металла и лигандов). В результате этого разность энергетических уровней d -орбиталей обычно ниже энергии спаривания электронов, и тетраэдрические комплексы обычно высокоспиновые.
Диаграммы распределения d -электронов позволяют предсказать магнитные свойства координационных соединений. Комплексы с непарными электронами являются парамагнитными и притягиваются магнитным полем, а без - диамагнитными и слабо отталкиваются.
Энергия стабилизации кристаллическим полем [ | ]
Энергия стабилизации кристаллическим полем (ЭСКП) - энергия электронной конфигурации иона переходного металла относительно средней энергии орбиталей. Стабилизация возникает вследствие того, что в поле лигандов энергетический уровень некоторых орбиталей ниже, чем в гипотетическом сферическом поле, в котором на все пять d -орбиталей действует одинаковая сила отталкивания, и все d -орбитали вырождены. Например, в октаэдрическом случае уровень t 2g ниже, чем средний уровень в сферическом поле. Следовательно, если в данных орбиталях находятся электроны, то ион металла более стабилен в поле лигандов относительно сферического поля. Наоборот, энергетический уровень орбиталей e g выше среднего, и электроны, находящиеся в них, уменьшают стабилизацию.
Энергия стабилизации октаэдрическим полем
В октаэдрическом поле три орбитали t 2g стабилизированы относительно среднего энергетического уровня на 2 / 5 Δ окт, а две орбитали e g дестабилизированы на 3 / 5 Δ окт. Выше были приведены примеры двух электронных конфигураций d 5 . В первом примере - низкоспиновый комплекс 3− с пятью электронами в t 2g . Его ЭСКП составляет 5 × 2 / 5 Δ окт = 2Δ окт. Во втором примере - высокоспиновый комплекс 3− с ЭСКП (3 × 2 / 5 Δ окт) − (2 × 3 / 5 Δ окт) = 0. В этом случае стабилизирующий эффект электронов в низкоуровневых орбиталях нейтрализуется дестабилизирующим эффектом электронов в высокоуровневых орбиталях.
Диаграммы расщепления d-уровня кристаллическим полем [ | ]
| октаэдрическая | пентагонально-бипирамидальная | квадратно-антипризматическая |
|---|---|---|
Теория кристаллического поля пришла на смену теории валентных связей в 40-х годах XX столетия. В чистом виде она сейчас не применяется, так как не может объяснить образование ковалентных связей в комплексных соединениях и совершенно не учитывает истинного состояния лигандов (например, их действительных размеров) даже в случае взаимодействий, близких к чисто электростатическим.
Уже с середины 50-х годов упрощенная теория кристаллического поля была заменена усовершенствованной теорией поля лигандов , учитывающей ковалентный характер химических связей между комплексообразователем и лигандом.
Однако наиболее общий подход к объяснению образования комплексных соединений дает теория молекулярных орбиталей (МО), которая в настоящее время превалирует над всеми остальными. Метод молекулярных орбиталей предусматривает и чисто электростатическое взаимодействие при отсутствии перекрывания атомных орбиталей, и всю совокупность промежуточных степеней перекрывания.
Рассмотрим основные понятия теории кристаллического поля , которая, как и теория валентных связей, все еще сохраняет свое значение для качественного описания химических связей в комплексных соединениях из-за большой простоты и наглядности.
В теории кристаллического поля химическая связь комплексообразователь – лиганд считается электростатической . В соответствии с этой теорией лиганды располагаются вокруг комплексообразователя в вершинах правильных многогранников (полиэдров ) в виде точечных зарядов . Реальный объем лиганда теорией во внимание не принимается.
Лиганды, как точечные заряды, создают вокруг комплексообразователя электростатическое поле (“кристаллическое поле”, если рассматривать кристалл комплексного соединения, или поле лигандов ), в котором энергетические уровни комплексообразователя и прежде всего d -подуровни расщепляются , и их энергия изменяется. Характер расщепления, энергия новых энергетических уровней зависит от симметрии расположения лигандов (октаэдрическое, тетраэдрическое или иное кристаллическое поле). Когда в качестве лигандов координируются молекулы H 2 O, NH 3 , CO и другие, их рассматривают как диполи , ориентированные отрицательным зарядом к комплексообразователю.
Рассмотрим случай октаэдрического расположения лигандов (например, -3 или 3+). В центре октаэдра находится ион-комплексообразователь М(+ n) с электронами на d -атомных орбиталях, а в его вершинах – лиганды в виде точечных отрицательных зарядов (например, ионы F - или полярные молекулы типа NH 3). В условном ионе М(+ n), не связанном с лигандами, энергии всех пяти d -АО одинаковы (т.е. атомные орбитали вырожденные ).
Однако в октаэдрическом поле лигандов d
-АО комплексообразователя попадают в неравноценное
положение. Атомные орбитали d
(z
2) и d(х 2 -у 2)
, вытянутые вдоль осей координат, ближе всего подходят к лигандам. Между этими орбиталями и лигандами, находящимися в вершинах октаэдра, возникают значительные силы отталкивания
, приводящие к увеличению энергии орбиталей. Иначе говоря, данные атомные орбитали подвергаются максимальному воздействию поля лигандов
. Физической моделью такого взаимодействия может служить сильно сжатая пружина.
Другие три d
-АО – d
(xy
), d
(xz
) и d
(yz
), расположенные между осями координат и между лигандами, находятся на более значительном расстоянии от них. Взаимодействие таких d
-АО с лигандами минимально, а следовательно – энергия d
(xy
), d
(xz
) и d
(yz
)-АО понижается по сравнению с исходной.
Таким образом, пятикратно вырожденные d
-АО комплексообразователя, попадая в октаэдрическое поле лигандов
, подвергаются расщеплению
на две группы новых орбиталей – трехкратно вырожденные орбитали
с более низкой энергией, d
(xy
), d
(xz
) и d
(yz
), и двукратно вырожденные орбитали
с более высокой энергией, d
(z
2) и d(х 2 -у 2)
. Эти новые группы d
-орбиталей с более низкой
и более высокой энергией
обозначают d
ε и d
γ:
d
(z
2) и d(х 2 -у 2)
d (xy ), d (xz ),d (yz )
Разность энергий двух новых подуровней d ε и d γ получила название параметра расщепления Δ 0:
E 2 – E 1 = Δ 0 ≈ 0
Расположение двух новых энергетических подуровней d ε и d γ по отношению к исходному (d -АО) на энергетической диаграмме несимметричное :
(Е 2 – Е 0) > (Е 0 – Е 1).
Квантово-механическая теория
требует, чтобы при полном заселении новых энергетических уровней электронами общая энергия осталась без изменения
, т.е. она должна остаться равной
Е
0 .
Иначе говоря, должно выполняться равенство
4(Е 2 – Е 0) = 6(Е 0 – Е 1),
где 4 и 6 – максимальное число электронов на d γ- и d ε-АО. Из этого равенства следует, что
(Е
2 – Е
0) / (Е
0 – Е
1) = 3/2 и
(Е
2 – Е
1) / (Е
0 – Е
1) = 5/2, или
Δ 0 / (Е 0 – Е 1) = 5/2, откуда (Е 0 – Е 1) = 2/5Δ 0 .
Размещение каждого электрона из шести максимально возможных на d ε-орбитали вызывает уменьшение (выигрыш ) энергии на 2/5 Δ 0 .
Наоборот, размещение каждого электрона из четырех возможных на d γ-орбитали вызывает увеличение (затрату ) энергии на 3/5 Δ 0 .
Если заселить электронами d ε- и d γ-орбитали полностью, то никакого выигрыша энергии не будет (как не будет и дополнительной затраты энергии ).
Но если исходная d
-АО заселена только частично
и содержит от 1 до 6 электронов, и эти электроны размещаются только на d
ε-АО, то мы получим значительный выигрыш энергии
.
Выигрыш энергии за счет преимущественного заселения
электронами d
ε-атомных орбиталей называют энергией стабилизации комплекса полем лигандов
.
Специфика каждого из лигандов сказывается в том, какое поле данный лиганд создает – сильное или слабое . Чем сильнее поле лигандов, чем больше значение параметра расщепления Δ 0 .
Изучение параметра расщепления, как правило, основано на спектроскопических исследованиях. Длины волн полос поглощения комплексов в кристаллическом состоянии или в растворе, обусловленные переходом электронов с d ε- на d γ-АО, связаны с параметром расщепления Δ 0 следующим образом:
λ = c / ν; Δ 0 = Е 2 – Е 1 = h ν = h · (c / λ),
где постоянная Планка h
равна 6,6260693 ∙ 10 -34 Дж · с;
скорость света с
= 3 · 10 10 см/с.
Единица измерения
Δ 0 – та же, что у волнового числа: см -1 , что приближенно отвечает 12 Дж/моль. Параметр расщепления
, помимо типа лиганда, зависит от степени окисления
и природы
комплексообразователя.
В комплексных соединениях, включающих комплексообразователи одного и того же периода и в одинаковой степени окисления, с одними и теми же лигандами, параметр расщепления примерно одинаков. С ростом степени окисления комплексообразователя значение Δ 0 увеличивается
. Так, для аквакомплексов 2+ и 2+ значение параметра расщепления составляет 7800 и 10400 см -1 , а для 3+ и +3 13700 и 21000 см -1 соответственно. При увеличении заряда ядра
атома-комплексообразователя Δ 0 тоже растет. Катионы гексаамминкобальта(III) 3+ , гексаамминродия(III) 3+ , гексаамминиридия(III) 3+ (Z
= 27, 45 и 77) характеризуются параметрами расщепления, равными 22900, 34100 и 41000 см -1 .
Зависимость Δ 0 от природы лигандов более разнообразна. В результате исследования многочисленных комплексных соединений было установлено, что по способности увеличивать параметр расщепления металлов-комплексообразователей, находящихся в своих обычных степенях окисления, наиболее распространенные лиганды можно расположить в следующий спектрохимический ряд
, вдоль которого значение Δ 0 монотонно растет:
I > Br > Cl > NCS - ≈ NO 3 - > F - > OH - >H 2 O > H - > NH 3 > NO 2 - > CN - > NO > CO.
Таким образом, наиболее сильное электростатическое поле вокруг комплексообразователя и самое сильное расщепление d -АО вызывают лиганды CN - , NO и CO. Рассмотрим распределение электронов по d ε- и d γ-орбиталям в октаэдрическом поле лигандов. Заселение d ε- и d γ-орбиталей происходит в полном соответствии с правилом Гунда и принципом Паули . При этом независимо от значения параметра расщепления первые три электрона занимают квантовые ячейки d ε-подуровня:
![]() dε
dε
Если число электронов на d -подуровне комплексообразователя больше трех, для размещения их по расщепленным подуровням появляется две возможности. При низком значении параметра расщепления (слабое поле лигандов) электроны преодолевают энергетический барьер, разделяющий d ε- и d γ-орбитали; четвертый, а затем и пятый электроны заселяют квантовые ячейки d γ-подуровня.
![]() dε
dε
При сильном поле лигандов и высоком значении Δ 0 заселение четвертым и пятым электроном d γ-подуровня исключено; происходит заполнение d ε-орбиталей.
![]() dε
dε
При слабом поле лигандов заселяющие квантовые ячейки 4 или 5 электронов имеют параллельные спины , поэтому получаемый комплекс оказывается сильно парамагнитен . В сильном поле лигандов образуются одна, а затем две электронные пары на d ε-подуровне, так что парамагнетизм комплекса оказывается гораздо слабее. Шестой, седьмой и восьмой электроны в случае слабого поля оказываются снова на d γ-подуровне, дополняя конфигурации до электронных пар (одной в случае d 6 , двух – d 7 и трех – d 8):
![]() dε
dε
В случае сильного поля лигандов шестой электрон заселяет dε -АО, приводя к диамагнетизму комплекса, после чего седьмой и восьмой электроны поступают на d γ-подуровень:
![]() dε
dε
Очевидно, при восьмиэлектронной конфигурации различия в строении между комплексами с лигандами слабого и сильного поля исчезают . Заселение орбиталей девятым и десятым электроном также не различается для комплексов обоих типов:
![]() dε
dε
Вернемся к рассмотрению электронного строения октаэдрических комплексных ионов 3+ и -3 . В соответствии с расположением в спектрохимическом ряду , аммиак NH 3 относится к числу лигандов сильного поля , а фторид-ион F - – слабого поля . В анионе -3 лиганды F - создают слабое кристаллическое поле (Δ 0 = 13000 cм -1), и все электроны исходной 3d 6 -АО размещаются на d ε- и d γ-орбиталях без какого-либо спаривания. Комплексный ион является высокоспиновым и содержит четыре неспаренных электрона, поэтому он парамагнитен :

В ионе 3+ лиганды NH 3 создают сильное кристаллическое поле (Δ 0 = 22900 см -1), все 3d 6 -электроны размещаются на более энергетически выгодной d ε-орбитали. Переход электронов с d ε- на d γ-орбитали невозможен из-за слишком высокого энергетического барьера . Поэтому данный комплексный катион является низкоспиновым , он не содержит неспаренных электронов и диамагнитен :

Аналогичным образом могут быть представлены схемы распределения электронов по орбиталям в октаэдрическом поле для ионов 2+ и -4:


Лиганды H 2 O создают слабое поле; обмен электронами между d
ε- и d
γ-орбиталями не вызывает затруднений и поэтому число неспаренных электронов в комплексном ионе такое же, как и в условном ионе Fe + II . Получаемый аквакомплекс – высокоспиновый, парамагнитный
.
Наоборот, лиганды CN - вызывают значительное расщепление d
-АО, составляющее 33000 см -1 . Это значит, что существует сильная тенденция к размещению всех электронов
на d
ε-орбиталях. Выигрыш энергии
, получаемый при таком заселении орбиталей, много больше энергетических затрат, обусловленных спариванием электронов.
C позиции метода валентных связей в гибридизации валентных орбиталей, образующих связь в аквакомплексе участвуют d -АО внешнего подуровня (4sp 3 d 2), а в низкоспиновом ― d -АО внутреннего подуровня (3d 2 4sp 3).
Таким образом, в высокоспиновых комплексах с лигандами слабого поля осуществляется гибридизация с участием d -АО внешнего подуровня, а низкоспиновых с лигандами сильного поля ― d -АО внутреннего подуровня. Количество неспаренных электронов в комплексе возможно определить методом электронного парамагнитного резонанса (ЭПР). С помощью приборов данного метода, называемых ЭПР спектрометрами, исследуются парамагнитные вещества.
Теория кристаллического поля позволяет объяснить появление той или иной окраски у комплексных соединений. Среди комплексных соединений значительное количество в кристаллическом состоянии и водном растворе отличаются яркой окраской. Так, водный раствор, содержащий катионы 2+ , окрашен в интенсивно синий цвет, катионы 3+ придают раствору фиолетовую окраску, а катионы 2+ красную. Если через раствор или кристаллический образец вещества пропускать свет видимой части спектра , то в принципе возможны три варианта физического поведения образца: отсутствие поглощения света любой длины волны (образец вещества бесцветен , хотя может иметь полосы поглощения в ультрафиолетовой области спектра); полное поглощение света во всем интервале длин волн (образец будет казаться черным ); наконец, поглощение света только определенной длины волны (тогда образец будет иметь цвет, дополнительный к поглощенному узкому участку спектра).

Таким образом, цвет раствора или кристаллов определяется частотой полос поглощения видимого света. Поглощение квантов света комплексами (например, имеющими октаэдрическое строение) объясняется взаимодействием света с электронами, находящимися на d ε-подуровне, сопровождаемое их переходом на вакантные орбитали d γ-подуровня. Например, при пропускании света через водный раствор, содержащий катионы гексаакватитана(III) 3+ , обнаруживается полоса поглощения света в желто-зеленой области спектра (20300 см -1 , λ=500 нм). Это связано с переходом единственного электрона комплексообразователя с d ε-АО на d γ-подуровень:

Поэтому раствор, содержащий 3+ , приобретает фиолетовый цвет (дополнительный к поглощенному желто-зеленому). Раствор соли ванадия Cl 3 имеет зеленый цвет. Это также обусловлено соответствующими переходами электронов при поглощении ими части энергии светового луча. В основном состоянии, при электронной конфигурации ванадия(III) 3d 2 , два неспаренных электрона занимают d ε-подуровень:

Существует всего два варианта перехода двух электронов
на d
γ-подуровень: либо оба
электрона занимают d
γ-АО, либо только один
из них. Любые другие переходы электронов, связанные с уменьшением суммарного спина, запрещены.
Указанным переходам электронов, получивших избыточную энергию, соответствует полоса поглощения
около 400 нм в спектре поглощения раствора хлорида гексаакваванадия(III). Поглощение пурпурно-фиолетовой области спектра дает дополнительный цвет раствора – ярко-зеленый
. Если комплексообразователь имеет электронную конфигурацию d
0 или d
10 , то переходы электронов
с d
ε- на d
γ-подуровень или наоборот невозможны
либо из-за отсутствия электронов
, либо из-за отсутствия вакантных орбиталей
. Поэтому растворы комплексов с такими комплексообразователями, как Sc(III) (3d
0), Cu(I) (3d
10), Zn(II) (3d
10), Cd(II) (4d
10) и т.п., не поглощают энергии в видимой части спектра и кажутся бесцветными
. Избирательность поглощения света зависит не только от комплексообразователя
и степени его окисления
, но и от вида лигандов
. При замене в комплексном соединении лигандов, находящихся в левой части спектрохимического ряда, на лиганды, создающие сильное
электростатическое поле, наблюдается увеличение
доли энергии, поглощаемой электронами из проходящего света и как следствие – уменьшение
длины волны соответствующей полосы поглощения. Так, водный раствор, содержащий катионы тетрааквамеди(II) 2+ , окрашен в голубой цвет, а раствор сульфата тетраамминмеди(II) 2+ имеет интенсивно синюю окраску.
Похожая информация.
